Prerequisites
Resources
Files for this walkthrough
The package containing the files for this walkthrough are available for download through the "Annotation of Conserved Motifs in Drosophila" page on the GEP website.
Introduction
The D. melanogaster Muller F element is unusual because it appears to be packaged as heterochromatin, but the genes that reside in this domain exhibit expression patterns that are similar to euchromatic genes [Riddle et al., 2012]. This dichotomy suggests that Muller F element genes either have some unique characteristics, or they have a more robust version of the gene activation machinery that enables them to function in a heterochromatic environment. For example, Muller F element genes might have distinct transcription factor binding sites (TFBS) near the transcription start sites (TSS) or they could simply have more enhancers around the TSS that facilitate the activation of a gene.
To begin to explore these hypotheses, the GEP Muller F element motif project will tabulate the types and the locations of known TFBSs within 2kb of the TSS of Muller F and Muller D element genes in multiple Drosophila species. Comparison of the motifs found in these two regions might identify motifs that are either enriched or depleted in Muller F element genes.
In this walkthrough, we will illustrate the protocol for annotating conserved motifs by searching for TFBSs near the promoter of onecut in the D. biarmipes project contig35 [Aug. 2013 (GEP/Dot) assembly]. Because of the lack of experimental data for D. biarmipes, we can only infer the locations of TFBSs based on sequence conservation with D. melanogaster.
Identify the putative transcription start site of onecut
Before we can search for TFBSs, we need to annotate the coding exons and identify the putative TSS of the onecut gene. The TSS annotation protocol is described in the "Annotation of Transcription Start Sites in Drosophila" walkthrough. In the current walkthrough, we assume that the annotator has already produced the gene annotations for onecut on the D. biarmipes Muller F element project contig35. The onecut gene is on the minus strand on contig35 and the first transcribed exon (onecut:3) of the A isoform has been placed at 18924-21599. The TSS for both isoforms of onecut is placed at 21,599.
Identify the transcription factor binding sites in D. melanogaster
Similar to the annotation of the coding and untranslated regions, the first step of our analysis is to determine the list of TFBSs that are present in the D. melanogaster gene. The modENCODE project has produced a map of TFBSs for 38 site-specific transcription factors in D. melanogaster early embryos. To summarize the TFBS results, the modENCODE project has also defined a list of High Occupancy Target (HOT) regions based on the number and the proximity of TFBSs in a given genomic region [Nègre et al., 2011].
To view the list of HOT regions for the onecut gene, open a new web browser window and navigate to FlyBase. Enter "onecut" into the "Jump to Gene" (J2G) search box in the top navigation bar and then click "Go". Click on the "GBrowse" button under the "Genomic Location" section of the FlyBase Gene Report for onecut (Figure 1).
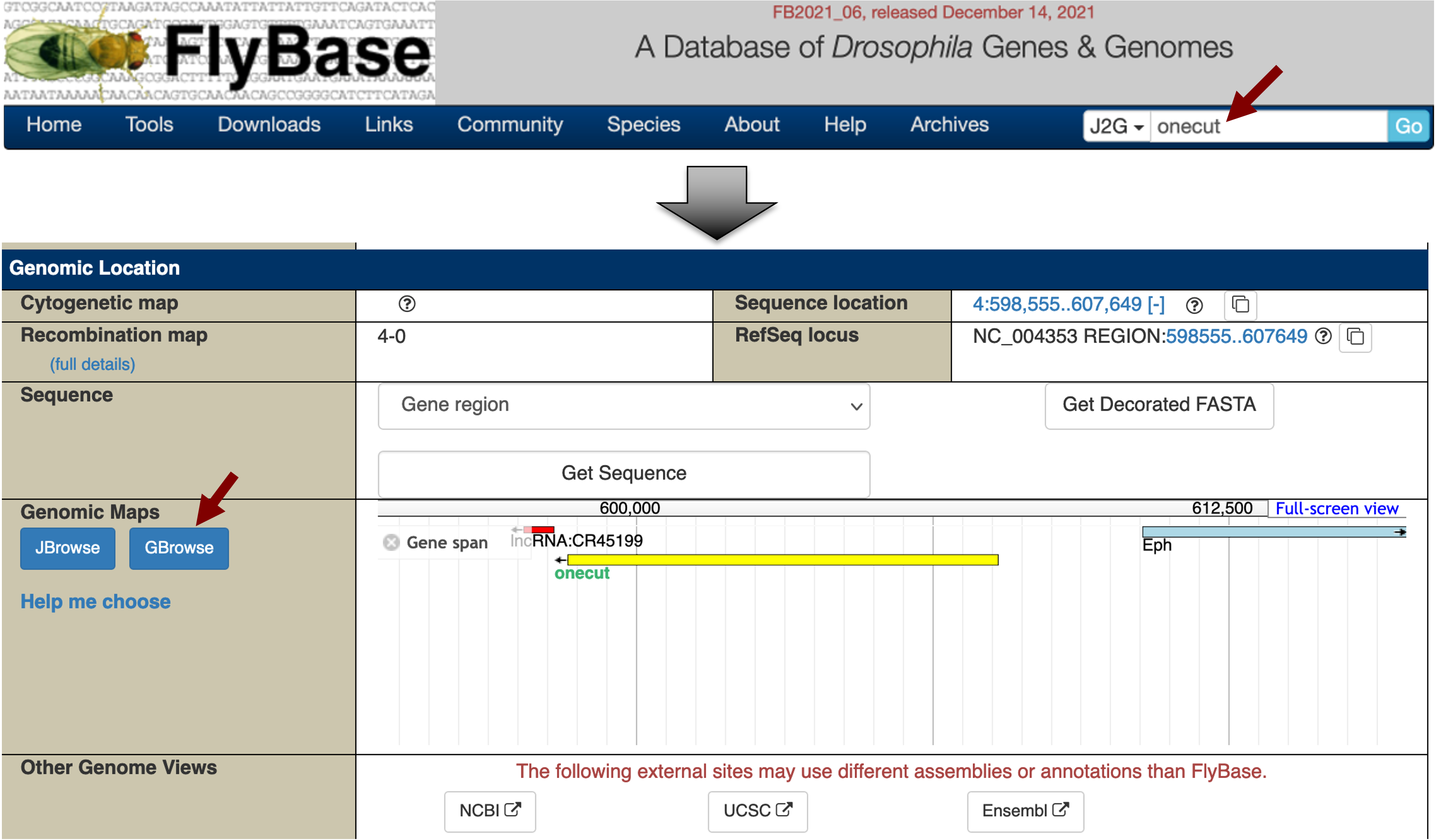
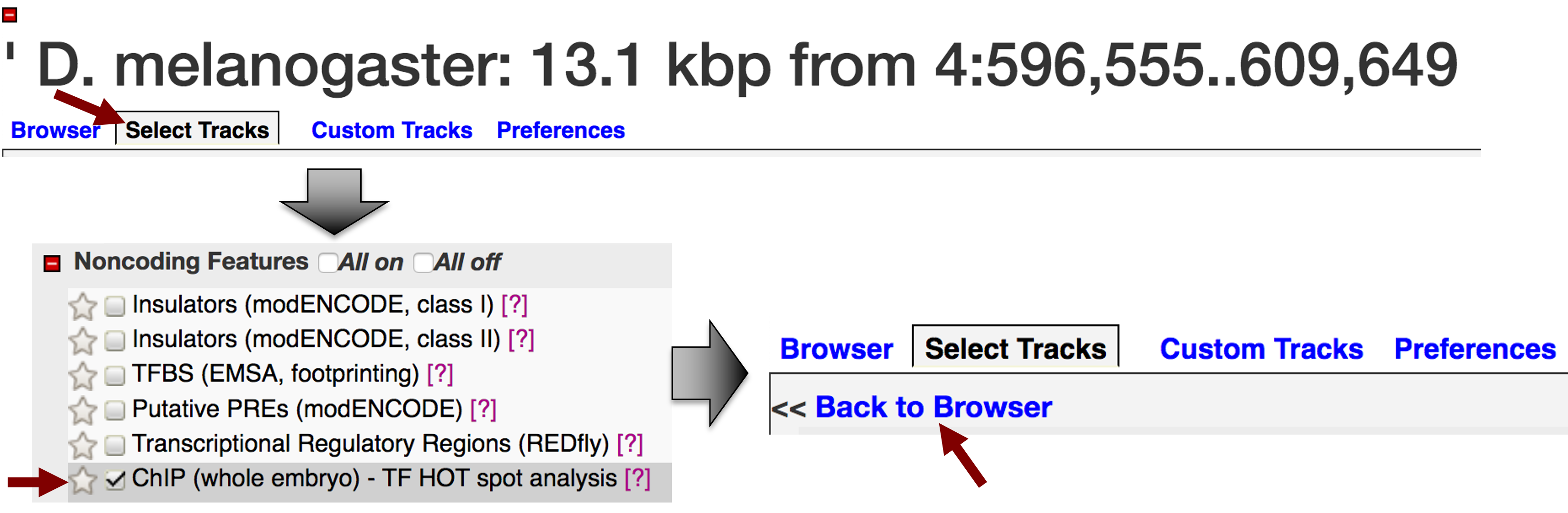
Click on the "Select Tracks" tab (next to the "Browser" tab). Scroll down and select the "ChIP (whole embryo) - TF HOT spot analysis" track under the "Noncoding Features" section (Figure 2). Click on the "Back to Browser" link at the top of the page (Figure 2) or the "Back to Browser" button at the bottom of the page to return to the graphical view of GBrowse.
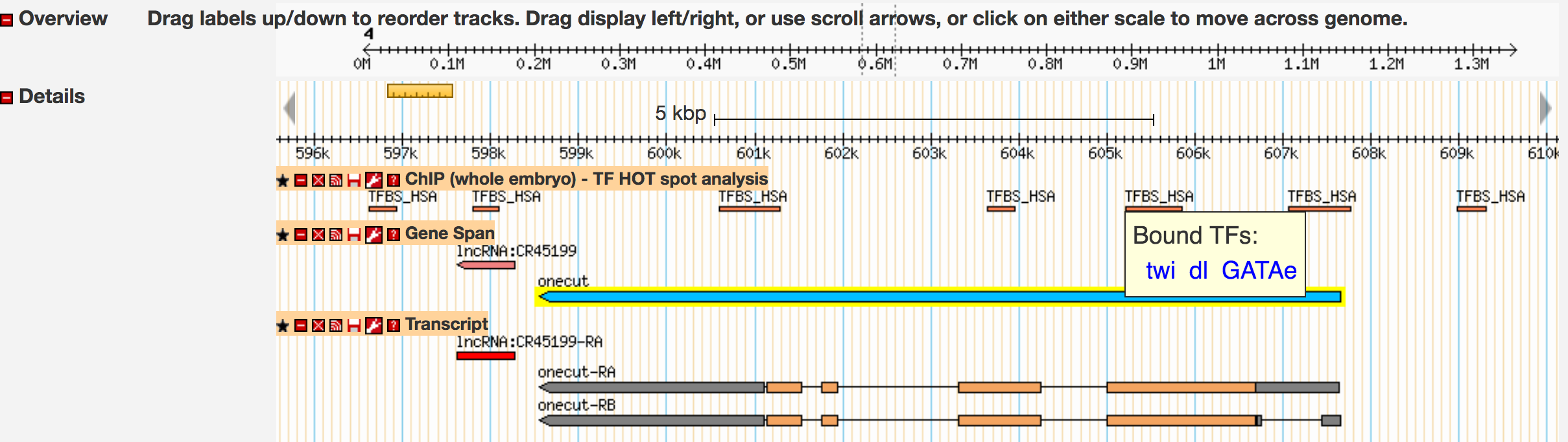
The "ChIP (whole embryo) - TF HOT spot analysis" track shows multiple HOT spots that overlap with the transcribed exons of the D. melanogaster onecut gene. There are also HOT spots in the regions immediately upstream and downstream of this gene. You can click on each of these HOT spots to view the complexity score and the transcription factors that are associated with each HOT spot (listed under Experimental Data → Binding data). The complexity score of a HOT spot is correlated with the distribution and the number of transcription factor binding sites (TFBS). A HOT spot with more TFBSs typically has a higher complexity score than a HOT spot with fewer TFBSs (see [Nègre et al., 2011]).
When we hover the mouse over the HOT spot that overlaps with the TSS of onecut (at ~607kb), a tooltip appears which indicates that this HOT spot is a binding site for the transcription factors GATAe, twist (twi), and dorsal (dl) (Figure 3). By contrast, the HOT spot at ~609kb is a binding site for only the transcription factor dl. We can click on the links within the tooltip to learn more about each transcription factor from the FlyBase Gene Report. In this walkthrough, we will only explore one of these transcription factors (dorsal) in more detail. In your own projects, you should analyze all the transcription factors that are associated with each HOT spot.
Using FlyFactorSurvey to determine the motif for the TFBS
Examination of the two HOT spots surrounding the TSS of onecut [one that overlaps with the 5' untranslated region (UTR) of onecut and one upstream of the TSS (at ~609kb)] shows that they both contain binding sites for dl (dorsal). To test whether these binding sites also exist in the D. biarmipes ortholog, we need to first determine the binding site motif for dl in D. melanogaster.
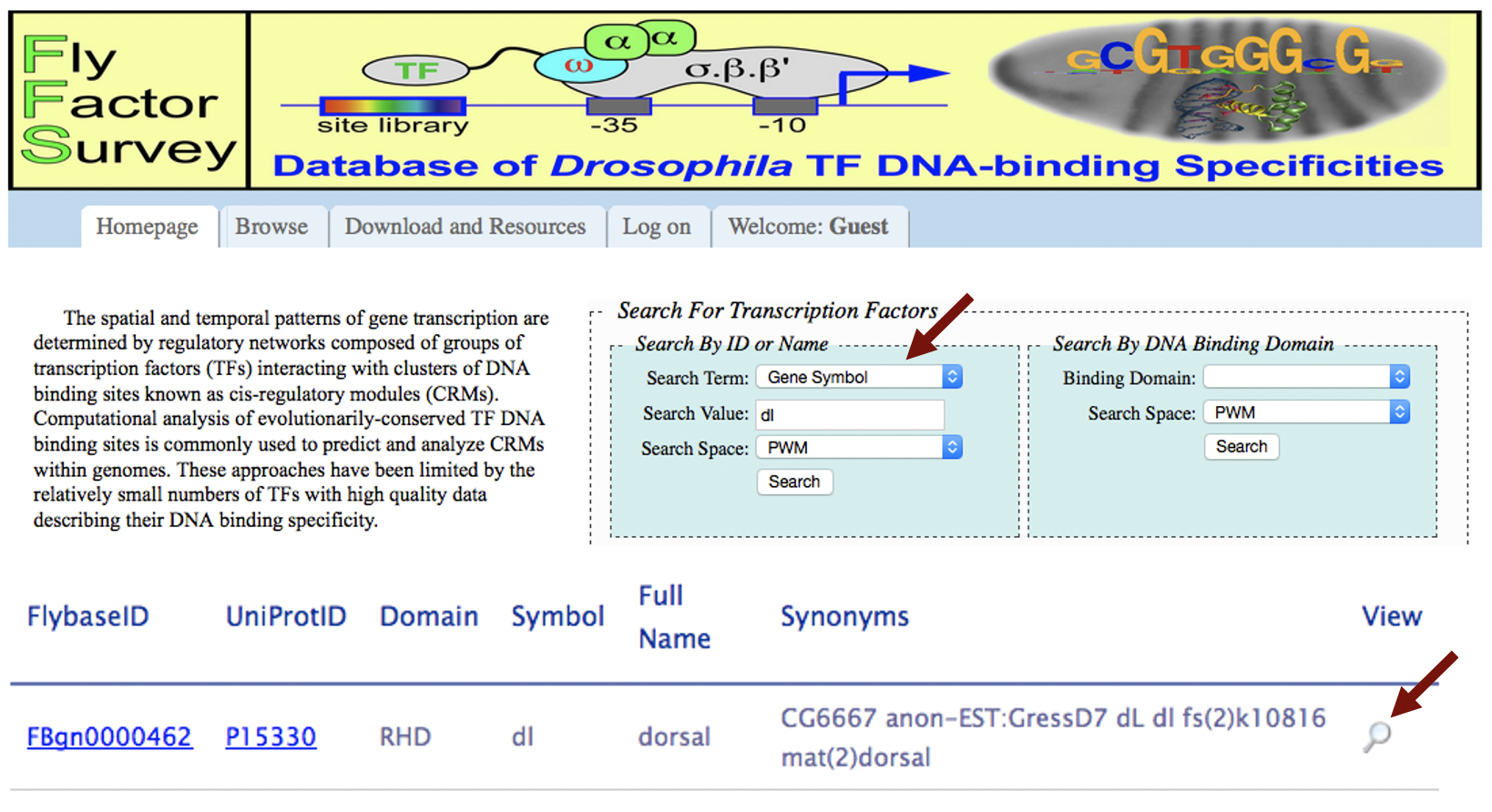
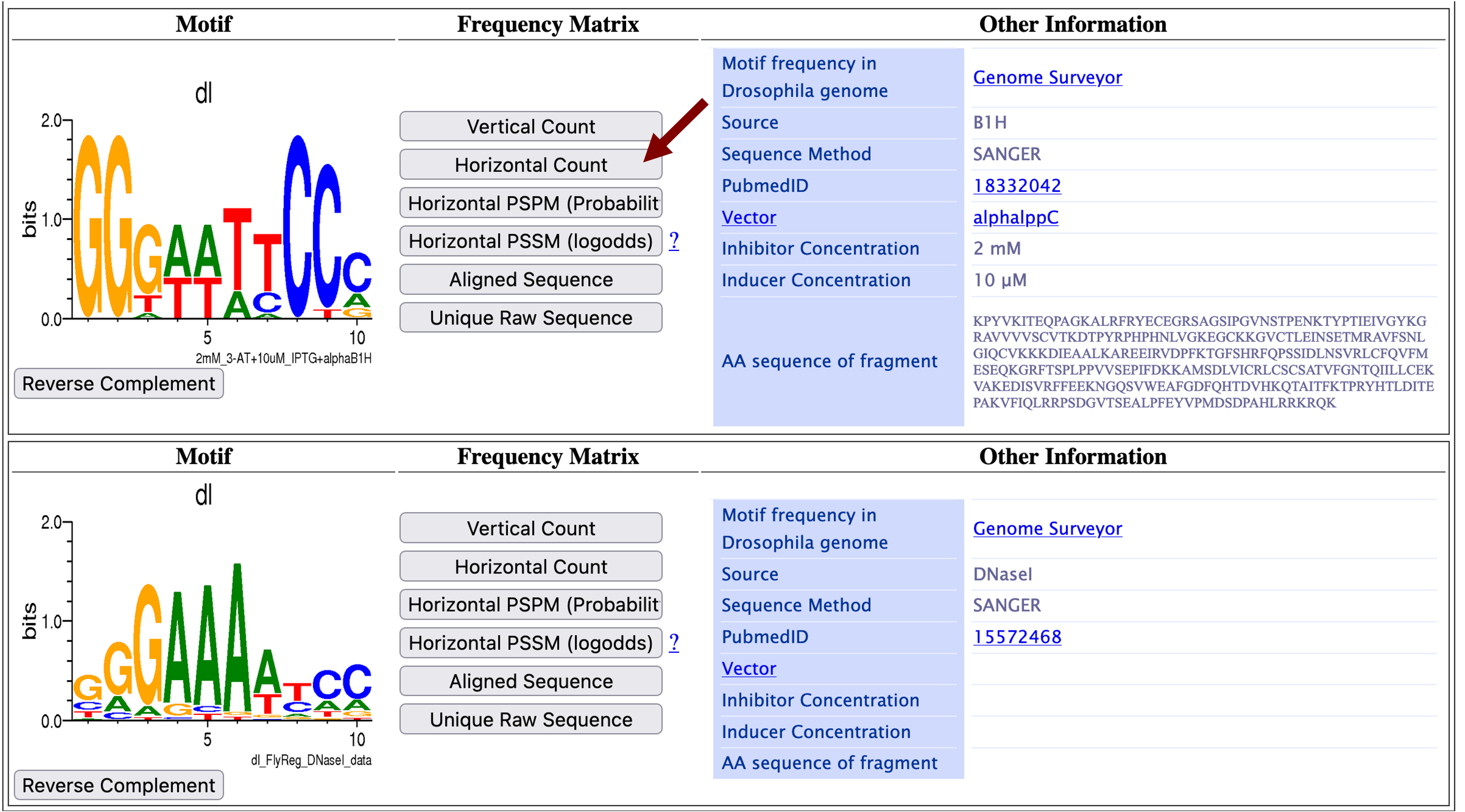
The FlyFactorSurvey database contains the binding site motifs for many transcription factors in D. melanogaster. These motifs have been experimentally determined using the bacterial one-hybrid method [Zhu et al., 2011]. To obtain the binding site motif for dl, navigate to the FlyFactorSurvey website. Under the "Search For Transcription Factors" section, change the "Search Term" field to "Gene Symbol" and the "Search Value" to "dl". Then click on the "Search" button. Click on the magnifying glass icon under the "View" column to view the record (Figure 4).
The Detail Viewer page shows two motifs for the TFBS of dl that were defined by two different studies. The "Source" field in the "Other Information" panel shows that the first motif is determined by the bacterial one-hybrid method (B1H) while the second motif is determined by DNase I footprinting. Each motif is shown as a sequence logo under the "Motif" column [Schneider and Stephens, 1990]. The sequence logo is constructed by aligning a collection of sequences (identified by B1H or DNase I, respectively) that contain a dl binding site. The x-axis corresponds to the aligned position of the nucleotides in the motif and the height of the letters correlate with the frequency of the nucleotide at each aligned position. For example, the sequence logo for the first dl motif (identified by B1H, Figure 5, top) shows that the first and second positions of the motif are always a G while the fourth and fifth positions have almost the same probability of being either an A or a T.
The total height of each column of the sequence logo corresponds to the information content (in bits) for that position. Since there are four nucleotides, the maximum amount of information for a position within a motif is two bits [i.e., log2(4)]. A motif position has two bits of information when all the motif instances used to construct the sequence logo have the same nucleotide at that position of the motif. The total information content of a motif corresponds to the sum of the information content for all the columns of a motif.
For the first dl motif, the sequence logo shows that the first position contributes ~1.8 bits of information (Figure 5, top). For the second dl motif, the sequence logo shows that the first position has lower information content than the first dl motif, as it contributes ~0.4 bits of information (Figure 5, bottom). When a motif is compared against a genome to identify potential binding sites, the alignment score for each position is weighted according to the information content of each column. For example, while the G nucleotide appears most frequently in the first column of both the first and second dl motifs, an alignment which contains a mismatch to the first column will incur a higher penalty for the first dl motif than then second dl motif.
Based on the total heights of the two sequence logos for dl, the motif defined by B1H (Figure 5, top) contains more information than the motif defined by DNase I (Figure 5, bottom). Since a motif with higher total information content produces fewer spurious matches than a motif with lower information content (i.e., has higher specificity), we will use the dl motif defined by B1H in this exercise.
We can retrieve the count matrix that was used to construct the sequence logo of the first dl motif by clicking on the "Horizontal Count" button (Figure 5). Save this file on your Desktop. The count matrix shows the frequency of each nucleotide at each position of the motif. Depending on your web browser settings, the file might be saved automatically in your Downloads folder. (For teaching purposes, we have included the matrix file UmassPGFE_PWMfreq_dl_NBT_Horizontal.txt in the exercise package.)
Using Patser to search for TFBS in D. biarmipes
Because the sequence logo shows that the dl TFBS consists of a short and degenerate sequence, we cannot use blastn to find the putative binding sites for dl in our D. biarmipes project. In addition, we will find many spurious matches in our search results when searching for TFBSs because short sequences are more likely to occur by chance. Hence we need to use a tool that can identify the subset of matches that are statistically significant.
We will use Patser to search for potential binding sites of dl in the region surrounding the TSS of the onecut ortholog in D. biarmipes. Because two of the dl binding sites overlap with the initial exon (i.e., onecut:3) of the A isoform of onecut in D. melanogaster, we will search for potential binding sites of dl in the region that encompasses the entire first transcribed exon to 2kb upstream of the putative TSS in our D. biarmipes ortholog (i.e., contig35:18924-23599). (Note that the search region is defined by the distribution of the TFBS HOT spots in the D. melanogaster ortholog so the search region will differ from gene to gene.)
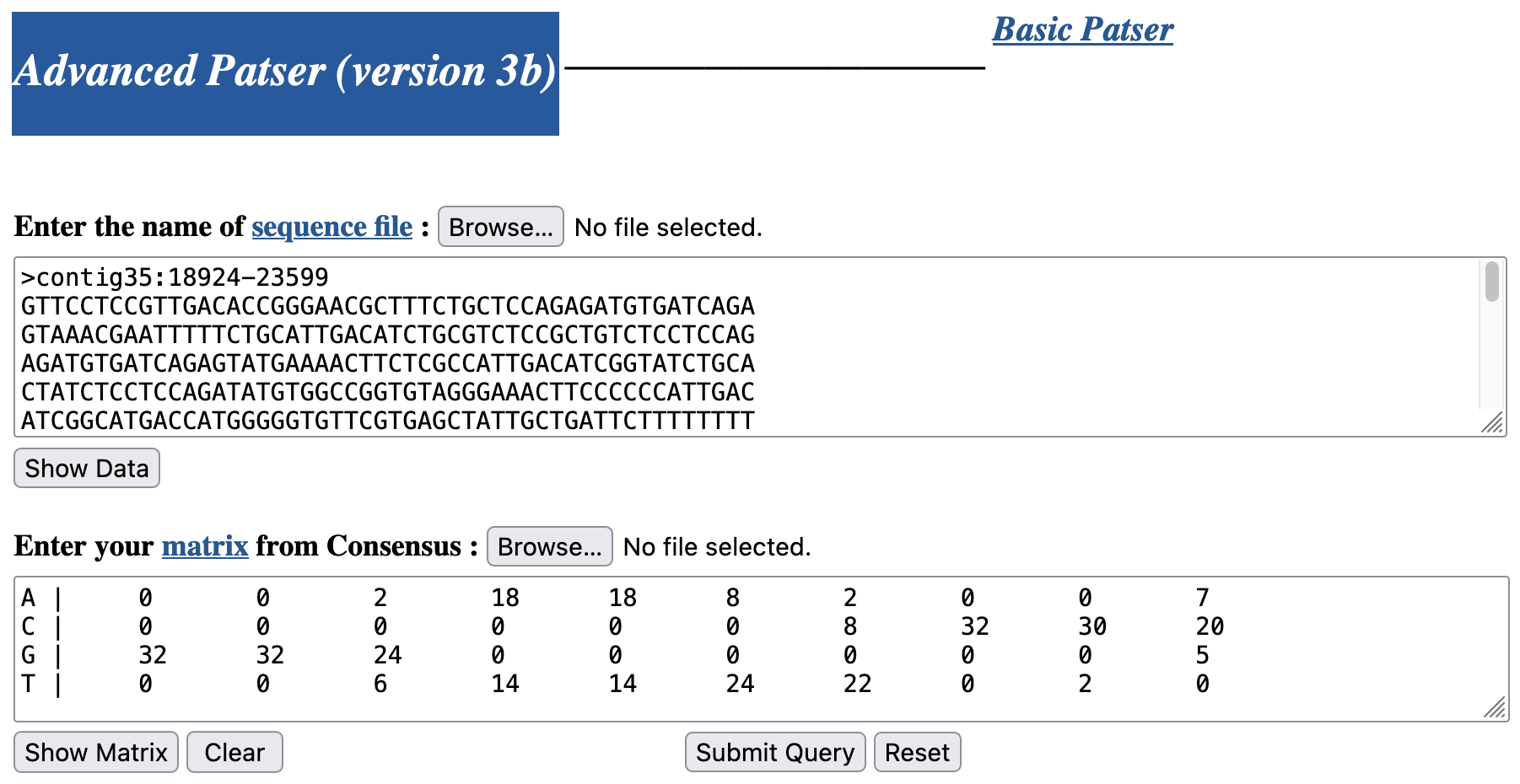
Open a new tab on your web browser and then navigate to the Consensus server. Click on the "Enter" link and then click on the "Advanced" link under the "Patser" section on the left navigation bar (Figure 6).

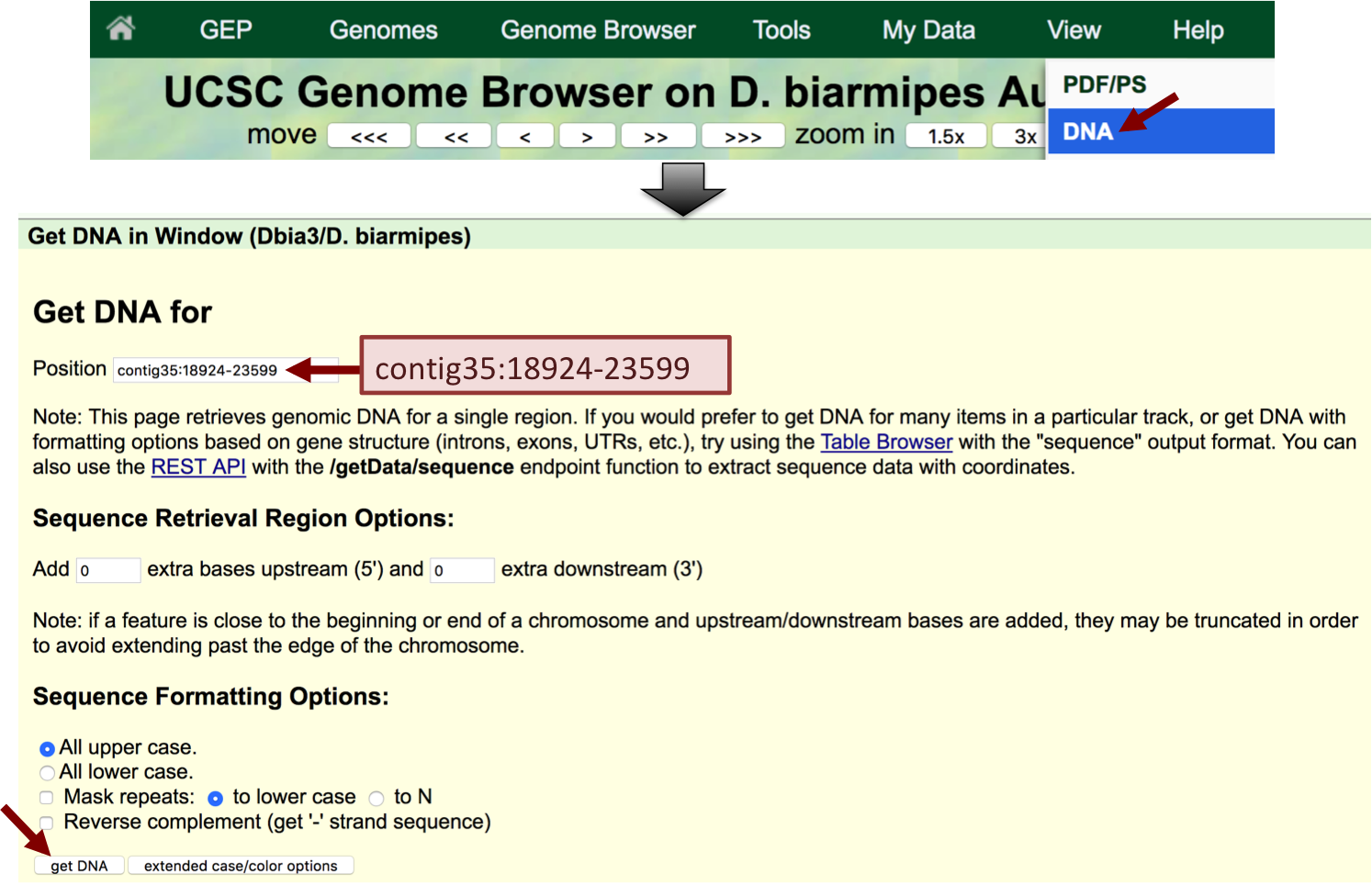
In order to perform the motif search with Patser, we need to specify the genomic sequence to search and the TFBS count matrix. We will use the GEP’s Mirror of the UCSC Genome Browser to retrieve the genomic sequence surrounding the onecut gene in D. biarmipes. Open a new tab and then navigate to GEP’s Mirror of the UCSC Genome Browser. Click on the "Genome Browser" link. Enter "D. biarmipes" into the "Enter species or common name" field. Select "Aug. 2013 (GEP/Dot)" under the "D. biarmipes Assembly" field, and enter "contig35" into the "Position/Search Term" field (Figure 7). Click on the "Go" button.

Click on the "DNA" button (under "View") in the main navigation bar. Enter "contig35:18924-23599" into the "Position" field and then click on the "get DNA" button (Figure 8). "Select all" the sequence and copy the sequence onto the clipboard. (For teaching purposes, we have included the sequence file contig35_onecut_upstream.fasta in the exercise package.)
Go back to the Patser web browser window. Paste the D. biarmipes sequence into the "Enter the name of sequence file" text box. Change the definition line (i.e., the line that begins with '>' at the beginning of the sequence) to ">contig35:18924-23599". Open the matrix file (i.e., UmassPGFE_PWMfreq_dl_NBT_Horizontal.txt) we have saved earlier in a text editor (e.g., WordPad on MS Windows, TextEdit on macOS). Copy only the four lines that begin with a nucleotide (i.e., skip the definition line which begins with '>') and then paste it into the "Enter your matrix from Consensus" text box (Figure 9).
Scroll down to the "Alphabet Options" section. Because the statistical significance of a match depends on the overall sequence composition, we need to specify the background base frequencies when we run Patser. The base composition for the D. biarmipes genome assembly is 58.2% A+T and 41.8% G + C. As an approximation, we will enter "a:t 3 c:g 2" in the "Seq. Alphabet and Normalization Information" field (Figure 10). (These values correspond to 60.0% A+T and 40.0% G+C.)

Scroll down to the "Output Options" section. By default, Patser will report the match score for every nucleotide in the query sequence. To show only the subset of matches that have positive scores, we will change the "Lowest score to print (-l)" to 1. Because transcription factors could bind in either orientation relative to the TSS, we will also select the option to "Score complementary sequence" (Figure 11).

Click on the "Submit Query" button at the bottom of the page to run the search. (For teaching purposes, the output from Patser is also available in the file Patser_onecut_dl_Dbiarmipes.txt in the exercise package.)
Interpreting the Patser results
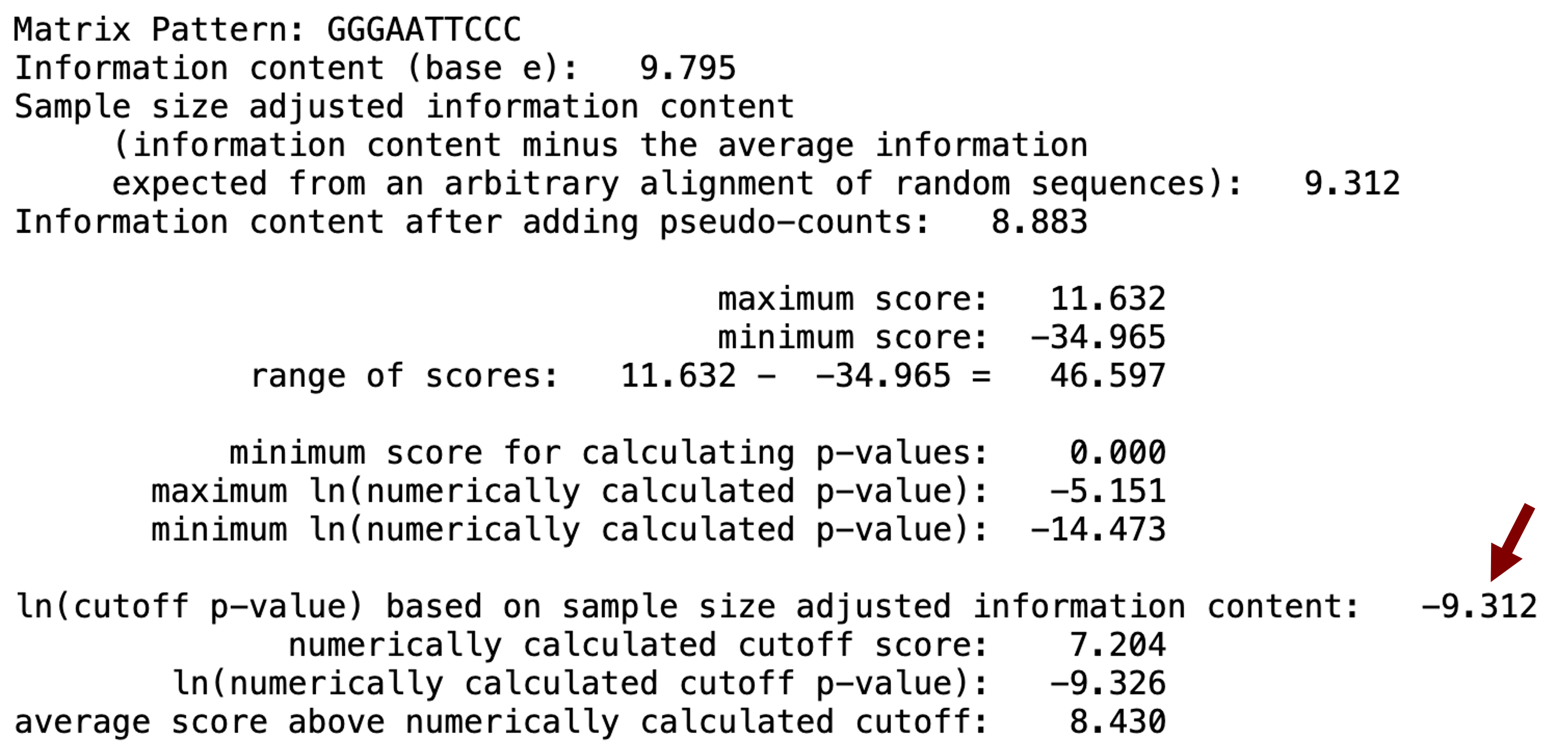
Part of the Patser output includes a suggested cutoff score for statistically significant motif matches. In this case, the suggested ln(cutoff p-value) after adjusting for the expected information content from an arbitrary alignment of random sequences (i.e., sample size) is -9.312 (Figure 12), which means that only matches with a ln(p-value) smaller (i.e., more negative) than -9.312 are significant. Among the 30 motif matches reported by Patser, only one match (at position 2597) has a ln(p-value) that is less than -9.312.
The suggested cutoff values calculated by Patser provide a good starting point for investigating potential candidate TFBS. However, the significance of a match depends on the level of motif conservation across different species, the degree of degeneracy of the binding site, and the size of the search region. Hence motif matches that are not statistically significant might nonetheless be biologically interesting.
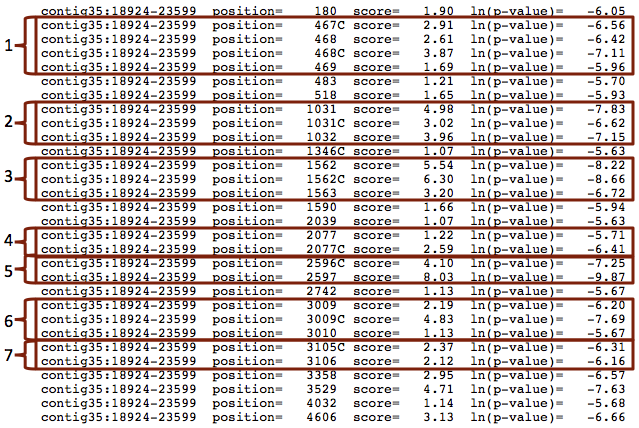
A closer examination of the Patser results shows that there are some positions within the search region that matched the dl TFBS motif on both the positive and negative strands. The "C" after the position denotes a match on the minus strand, i.e., the complement. We also find that some of the motif matches are located immediately adjacent to each other (Figure 13).
Many transcription factors bind to the DNA as either a homodimer (i.e., two of the same transcription factor) or heterodimer (i.e., two different transcription factors); previous studies have shown that dl binds to DNA as a homodimer [Govind et al., 1992]. Consequently, while the seven regions identified by Patser are not statistically significant, they still merit further investigation (e.g. via wet lab experiments) to determine if they correspond to functional binding sites of dl.
The positions reported by Patser are with respect to the extracted region and the motif of dl consists of 10 nucleotides. We can transform the coordinates reported by Patser so that they are with respect to the start of the contig35 sequence by adding 18,923 to all of the matched positions. For example, converting the position of the match at 2597 to the contig35 coordinate system (i.e., 2597+18924-1) means that the putative dl binding site begins at 21,520 in contig35.
However, because the motif match can be in either orientation, you must take the orientation of the motif match into account when you calculate the span of each motif match. (The Excel workbook "Patser_onecut_dl_Dbiarmipes.xlsx" in the exercise package contains additional details on how to transform the coordinates reported by Patser to the coordinates with respect to the entire contig35 sequence.) The list of putative binding sites for dl is summarized in the table below:
| Motif Candidate | Region |
|---|---|
1 |
19381-19401 |
2 |
19945-19964 |
3 |
20476-20495 |
4 |
20991-21009 |
5 |
21510-21529 |
6 |
21923-21942 |
7 |
22019-22038 |
Conclusions
This walkthrough illustrates how we can identify conserved TFBS in the putative ortholog of the onecut gene on the D. biarmipes Muller F element. Using FlyBase GBrowse, we examined the modENCODE HOT spot annotations near the TSS of the onecut gene in D. melanogaster. For each of the TFBS associated with the HOT spot, we can retrieve the corresponding horizontal count matrix from the FlyFactorSurvey database. We can then use this count matrix with Patser to determine if the TFBS is also present in the region surrounding the TSS of the D. biarmipes onecut ortholog.
This walkthrough illustrates how we can annotate the putative TFBS for dl in the region surrounding the TSS of the D. biarmipes onecut ortholog. We can apply this search strategy to the other TFBSs (i.e., twi, GATAe, and sens) that are found within the HOT spots that overlap with exon onecut:3 in D. melanogaster in order to determine if these binding sites are also conserved in D. biarmipes.
The comparative analysis so far suggests that the dl transcription factor likely plays an important role in regulating the transcription of the onecut gene in both D. melanogaster and D. biarmipes. However, we do not have any experimental data examining the evolution of the dl consensus binding site, which could differ in D. biarmipes, so this remains a hypothesis until further testing.
Bibliography
-
[Govind et al., 1992] Govind S., Whalen A. M., Steward R., 1992 In vivo self-association of the Drosophila rel-protein dorsal. Proc. Natl. Acad. Sci. U. S. A. 89: 7861–7865.
-
[Nègre et al., 2011] Nègre N., Brown C. D., Ma L., Bristow C. A., Miller S. W., et al., 2011 A cis-regulatory map of the Drosophila genome. Nature 471: 527–531.
-
[Riddle et al., 2012] Riddle N. C., Jung Y. L., Gu T., Alekseyenko A. A., Asker D., et al., 2012 Enrichment of HP1a on Drosophila chromosome 4 genes creates an alternate chromatin structure critical for regulation in this heterochromatic domain. PLoS Genet. 8: e1002954.
-
[Schneider and Stephens, 1990] Schneider T. D., Stephens R. M., 1990 Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 18: 6097–6100.
-
[Zhu et al., 2011] Zhu L. J., Christensen R. G., Kazemian M., Hull C. J., Enuameh M. S., et al., 2011 FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res. 39: D111–117.