Recommended background tutorial
An Introduction to NCBI BLAST
Files for this exercise
The package containing the files for this exercise is available through the “Detecting and Interpreting Genetic Homology” page on the GEP website.
Introduction
The Basic Local Alignment Search Tool (BLAST) is a program that reports regions of local similarity (at either the nucleotide or protein level) between a query sequence and sequences within a database. The ability to detect sequence homology allows us to determine if a gene or a protein is related to other known genes or proteins. Detecting sequence homology also facilitates the identification of conserved domains that are shared by multiple genes and the identification of members of a gene family.
BLAST is popular because it can efficiently identify regions of local similarity between two sequences. More importantly, BLAST is based on a robust statistical framework. This framework allows BLAST to determine if the alignment between two sequences is statistically significant (i.e., probability of obtaining an alignment with this score or higher by chance is low).
Before proceeding with annotation, it is important to understand the inferences that we are making when we use BLAST in our analysis. The theory of evolution proposes that all organisms descend by speciation from common ancestors. At the molecular level, an ancestral DNA sequence diverges over time (through accumulation of point mutations, duplications, deletions, transpositions, recombination events, etc.) to produce diverse sequences in the genomes of subsequent organisms. Mutations to sequences with an important biological function, such as genes, have a higher probability of being deleterious to the organism, so they are less likely to become fixed in a population. We say that such sequences are under negative selection, which causes them to be conserved against change over time. We therefore expect that two homologous copies of a functional sequence, either in two species or within a single species, will exhibit a higher degree of conservation, and therefore of base-by-base similarity, than either two unrelated sequences or two sequences that are not under strong negative selection. This similarity is the “signal” detected by a BLAST search.
When we perform a BLAST search, we reverse the above line of reasoning to infer common function from sequence similarity. We first use the observed sequence similarity to infer that two sequences are in fact conserved homologs. Then we use this inferred conservation to infer that the sequences have a common function (e.g., that they encode the same protein). There are, of course, limitations to this line of reasoning. For example, two unrelated sequences might appear similar purely by chance. Alternatively, a pair of sequences may be conserved homologs, but they may have diverged only recently (as in human and chimpanzee), so that conservation implies nothing about whether they are under negative selection. Finally, a pair of sequences may indeed be conserved homologs because of strong negative selection, yet have different functions. For example, the delta 1 and delta 2 crystallin genes in ducks have high sequence similarity and both serve structural functions in the eye lens. However, the delta 2 crystallin exhibits an additional enzymatic (arginosuccinate lyase) activity that is absent from the delta 1 crystallin. Similarly, the ADH1 and ADH2 genes in yeast are closely-related but have opposite enzymatic function! While BLAST is a powerful tool for detecting similarity, we must also understand its inherent limitations in order to properly interpret its results.
Overview of the annotation process
The main goal of this exercise is to identify interesting features (functional genes or non-functional pseudogenes) within a region of the D. melanogaster genome. We will use the programs BLAST and RepeatMasker to help us with the annotation. During the course of our investigation, we will also learn how to adjust some of the parameters in BLAST and in RepeatMasker to increase the sensitivity and specificity of our searches.
Much of this exercise consists of questions, which you should try to answer as you work through this exercise. You should also make note of the exact BLAST and RepeatMasker parameters and the databases that you use for your searches in order to ensure that your results are reproducible.
This exercise assumes that you are familiar with the basics of NCBI BLAST. You can find additional information on how to use BLAST at the NCBI BLAST Help page.
Finding interspersed repeats
The file dmel_seq1.fasta contains a FASTA-formatted DNA sequence, which represents roughly 4,000 bases from the X chromosome of D. melanogaster.
Before we attempt to search for genes in this 4kb sequence, we should first annotate its repetitive elements using RepeatMasker. Repetitive DNA elements are sequence motifs repeated hundreds or thousands of times in the genome, constitutes the major proportion of all the nuclear DNA in most eukaryotic genomes, and its significance is not fully understood. RepeatMasker is a program that identifies transposable elements and low complexity repeats in DNA sequences. You can run the RepeatMasker program at the RepeatMasker web server.
|
|
Alternatively, the result of the RepeatMasker analysis of our sequence is available in the exercise package (files within the DmelSeq1_RpM directory). |
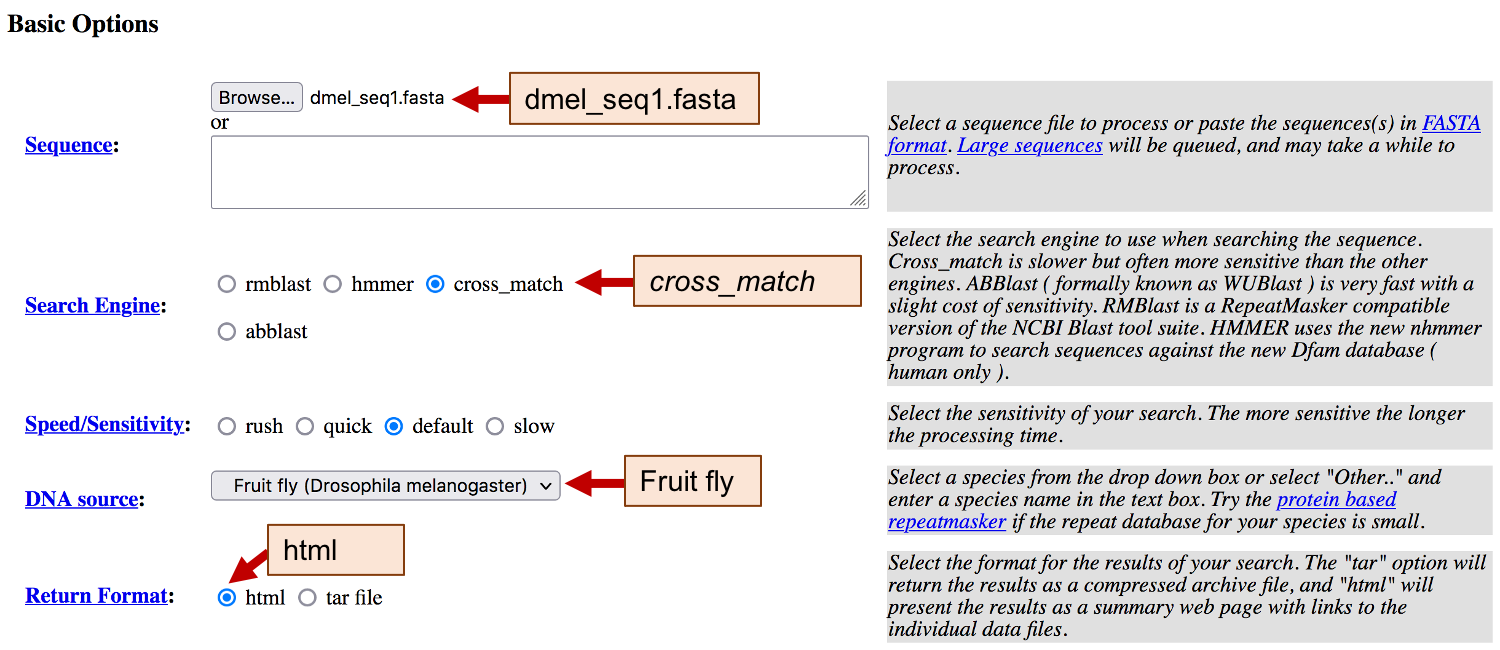
Open a new web browser window and navigate to the RepeatMasker web service. Click on the “Browse” or “Choose File” button under the “Sequence” field, and then select the dmel_seq1.fasta file from the exercise package.
We can use four different search engines with RepeatMasker: rmblast, hmmer, cross_match, and abblast. By default, RepeatMasker uses the rmblast search engine, which is faster but less sensitive than the hmmer or cross_match search engines. Since RepeatMasker works by comparing the query sequence against a database of known repetitive elements, we should select the repeat database that best corresponds to the sequence we are annotating. After all, we would not want to waste time looking for Alu repeats in our fly sequence! Since our sequence is from a fruit fly, we will use the Drosophila repeat library. Click on the “DNA source” drop-down menu and change it to “Fruit fly (Drosophila melanogaster)”. Verify that the “Return Format” is set to “html” so that we can view the RepeatMasker results in the web browser (Figure 1).
By default, RepeatMasker also masks simple repeats and low complexity DNA. Low complexity DNA sequences may not have a highly repetitive structure, but these regions consist primarily of one or two out of the four possible nucleotides.
Why might it be a good idea to remove low-complexity DNA from a sequence before running blastn? Why might it be a bad idea to do so before running blastx?
|
|
Consider proteins such as collagen that have highly regular sequences. |
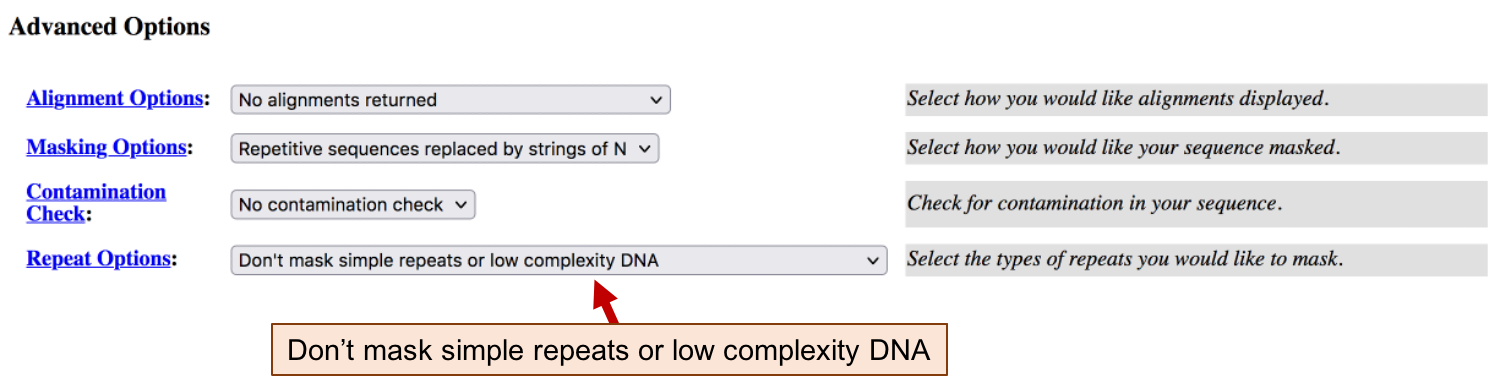
Because BLAST automatically filters low complexity regions when appropriate, we will tell RepeatMasker not to mask low complexity regions (Figure 2). Under “Advanced Options” change “Repeat Options” to “Don’t mask simple repeats or low complexity DNA”. Click “Submit Sequence”. Depending on how busy the server is, this analysis may take a few minutes to complete.
|
|
For class purposes, the RepeatMasker result files are also available inside the folder DmelSeq1_RpM in the exercise package. |
|
|
Note that RepeatMasker will cache the results of previous RepeatMasker analyses. If you see a message indicating that "Your request was previously run" or "Your request is still in the queue as id -1," then click on the link to view the RepeatMasker results. |
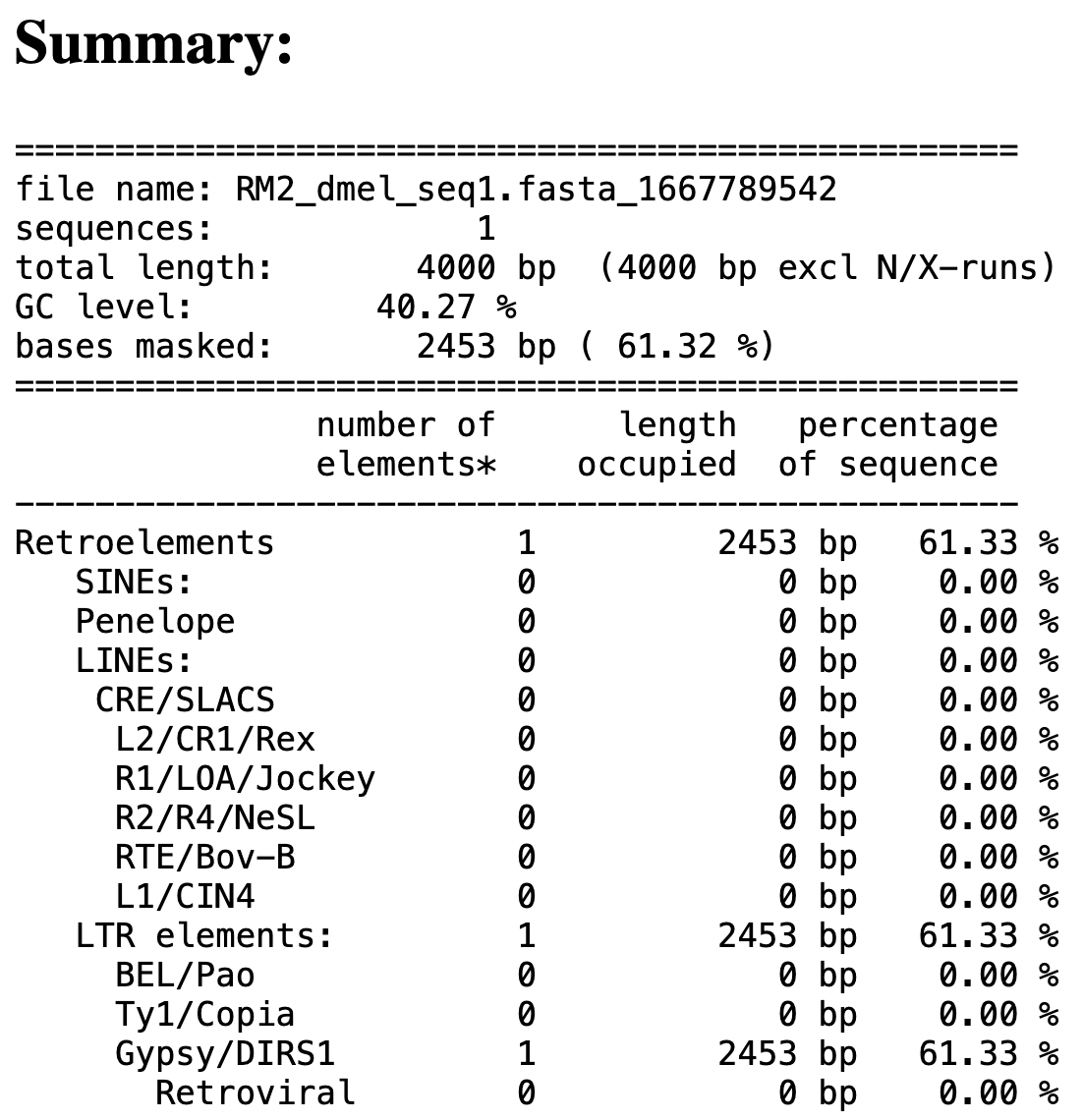
The results page shows a table that summarizes the types and number of repeats identified by RepeatMasker (Figure 3). Scroll down to the “Results” section to view or download the Annotation Files, Masked File, and Alignment File produced by RepeatMasker (Figure 4).
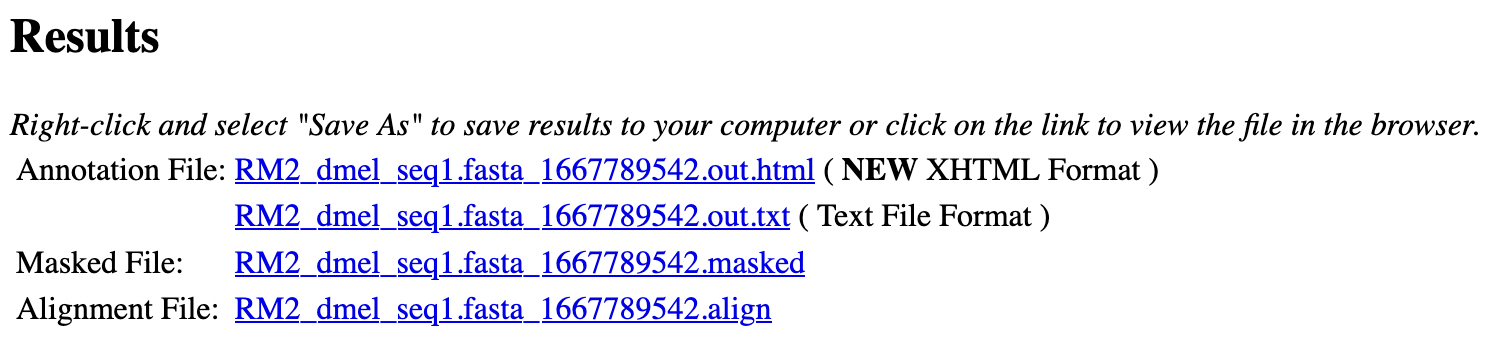
If RepeatMasker found repeats in the query sequence, then it will produce the following files:
-
A web page with a detailed list of repetitive elements found by RepeatMasker and their corresponding alignments, in a file with the extension “.out.html”
-
A plain text version of the list of repetitive elements found by RepeatMasker, in a file with the extension “.out.txt”
-
A copy of the original sequence with its repeats replaced by Ns, in a file with the extension “.masked”
-
A list of alignments of the query sequence against each repetitive element identified by RepeatMasker, in a file with the extension “.align”
How many repetitive elements does our sequence contain, and what are their types?
|
|
Examine the RepeatMasker Summary Table. |
In the next section, we will be working with another sequence, dmel_seq2.fasta. This 4.5kb sequence also comes from the X chromosome of D. melanogaster. Run RepeatMasker on this sequence using the same options as we have discussed above.
|
|
Alternatively, the RepeatMasker analysis of our sequence is available in the exercise package (within the DmelSeq2_RpM directory). |
What is your result? Given the length of this sequence, would you expect the same result if it had come from a primate?
Translated query vs. protein database (blastx): the gene hunter
Following the initial annotation of repetitive elements found within our sequence (dmel_seq2.fasta), we would like to see if any part of our sequence matches any known genes. We will use BLAST to help us detect these homologous regions.
There are a few decisions we must make before proceeding with the BLAST search. We could look for matches to our sequence at either the DNA or the protein level, using any one of several databases. In deciding which comparison tool to use, we should consider a few factors:
-
How sensitive will the comparison be? Is it likely to find genes or other meaningful features in our sequence?
-
How specific will the matches returned by our tool be? Will they cover the entire region or will they be confined to specific features of interest?
-
How good is the information associated with any matches we may find? Will we be able to interpret those matches?
-
How long will the tool take to run?
Considering all these factors, a reasonable first step to characterize anonymous DNA sequence is to compare the DNA sequence against the UniProtKB/Swiss-Prot protein database (a database of well characterized proteins) using blastx. In a blastx search, a nucleotide query sequence is translated into peptide sequences in all six reading frames (i.e., three reading frames on each strand) and compared against a protein database. This means blastx is good at specifically identifying parts of a DNA sequence that have the potential to code for proteins similar to those in the protein database. Hence it should provide us with a relatively good picture of potential genes (true positives) in our sequence without a lot of clutter (false positives).
If our blastx search against the UniProtKB/Swiss-Prot database fails to produce any significant matches, we could search our sequence against all proteins in the GenBank non-redundant (nr) protein database. The nr protein database contains most of the real and hypothetical peptide sequences that have been submitted to GenBank. While this would increase our chances of seeing a match, the quality of the supplementary information associated with each protein record in the nr database is much lower than those in the UniProtKB/Swiss-Prot database.
Note that there are also species-specific databases [e.g., FlyBase for fruit flies (Figure 5), WormBase for C. elegans] available on the web. Using these species-specific databases can substantially reduce the computational time needed to perform the BLAST searches. These species-specific databases might also contain additional metadata and references for each gene. However, we will use the generic databases in this exercise.
When performing BLAST searches, we will typically use the repeat-masked version of the sequence to reduce the search time and the number of spurious matches. However, because our sequence did not contain any repetitious elements, we will use the original sequence for our BLAST searches.
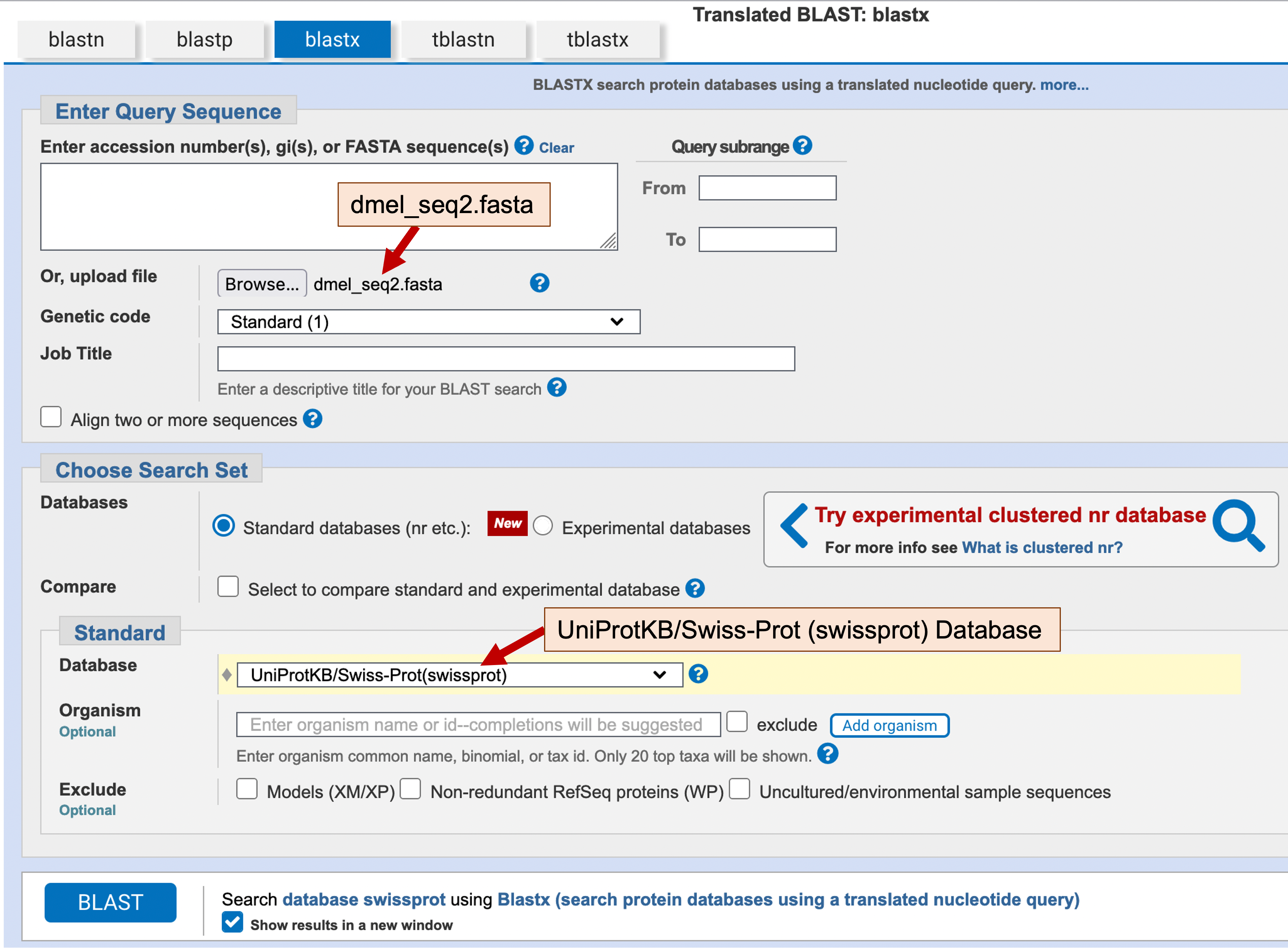
Open a new web browser window, navigate to the NCBI BLAST web server. Click on the blastx image under the “Web BLAST” section. Click on the “Browse” or “Choose File” button under the “Enter Query Sequence” section, and then select the dmel_seq2.fasta file from the exercise package. Under the “Database” menu, select “UniProtKB/Swiss-Prot (swissprot)” (Figure 6).
|
|
NCBI is constantly updating the databases and will occasionally change the default BLAST parameters. Hence you might not get exactly the same results if you were to run the searches yourself. You should run these searches to practice using these web pages. However, you may wish to use the results saved in the exercise package to answer the questions below. The results of this BLAST search are in the file “blx_swissprot_dmel_seq2.txt” inside the “blast_results” folder. |
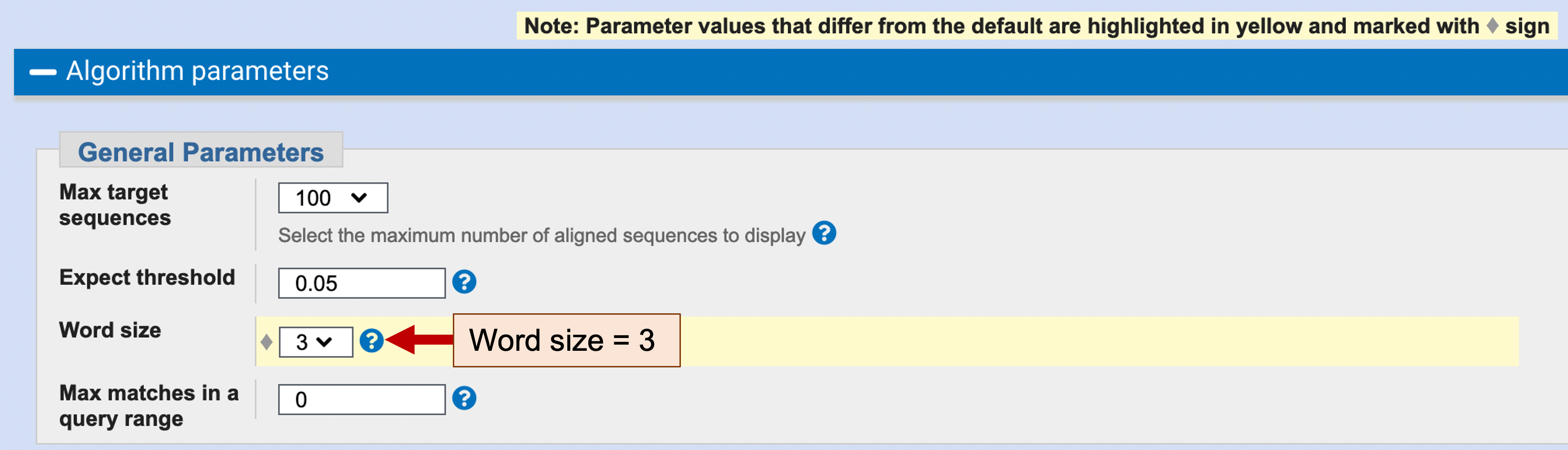
By default, blastx uses a Word size of 5 (i.e., a sliding window of 5 amino acids) to identify the positions within the query and subject sequences where blastx will initiate the local alignments. In order to increase the sensitivity of the blastx search, we will change the Word size to 3. Click on the “+” icon next to the “Algorithm parameters” header under the “BLAST” button to expand the section. Change the “Word size” parameter to 3 under the “General Parameters” section (Figure 7). Click on the “BLAST” button to run the blastx search.
How many blastx hits to distinct sequences were returned? What are the best and worst E-values reported?
When searching a large database, it is good practice to ignore matches with poor (high) E-values. In principle, a match with an E-value less than 1 is a significant hit (because we expect to find an alignment this good or better by chance less than once when searching the database). In practice, you should allow a large margin of safety when interpreting E-values because of the simplified model used by BLAST to calculate the alignments and E-values. As a rule of thumb, unless you are working with very short sequences, you should be suspicious of matches with E-values greater than 1e-10 and extremely skeptical of hits with E-values above 1e-5.
Based on the search result, what does BLAST say about the content of this sequence? What caveats might you consider in interpreting these results?
Interpreting the blastx output
If you emulated our analysis thus far, the hit table under the “Descriptions” tab should show two strong hits to the swallow protein (Figure 8). However, remember that sequence similarity does not necessarily imply that our sequence contains the D. melanogaster version of the swallow protein. We need to gather more evidence before deciding how to annotate our sequence.
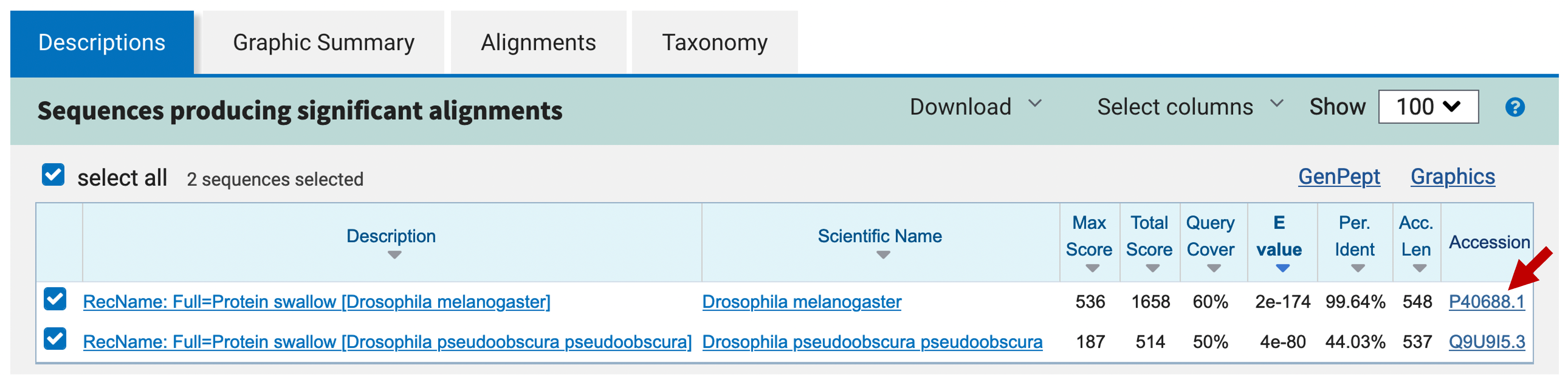
First, we need to find out more about the swallow protein. A good place to start is the UniProtKB/Swiss-Prot database, which is manually curated and has links to many other databases. To access the UniProtKB/Swiss-Prot record for the D. melanogaster swallow protein, click on the accession number “P40688.1” in the blastx hit table (red arrow, Figure 8). This will bring you to the GenBank record for the swallow protein (Figure 9). According to the GenBank record, the locus name (i.e., the UniProtKB accession string) for the swallow protein is SWA_DROME (blue arrow, Figure 9). A UniProtKB accession string consists of an abbreviated gene name followed by an abbreviated species name. For example, the name SWA_DROME corresponds to the swallow protein from DROsophila MElanogaster.
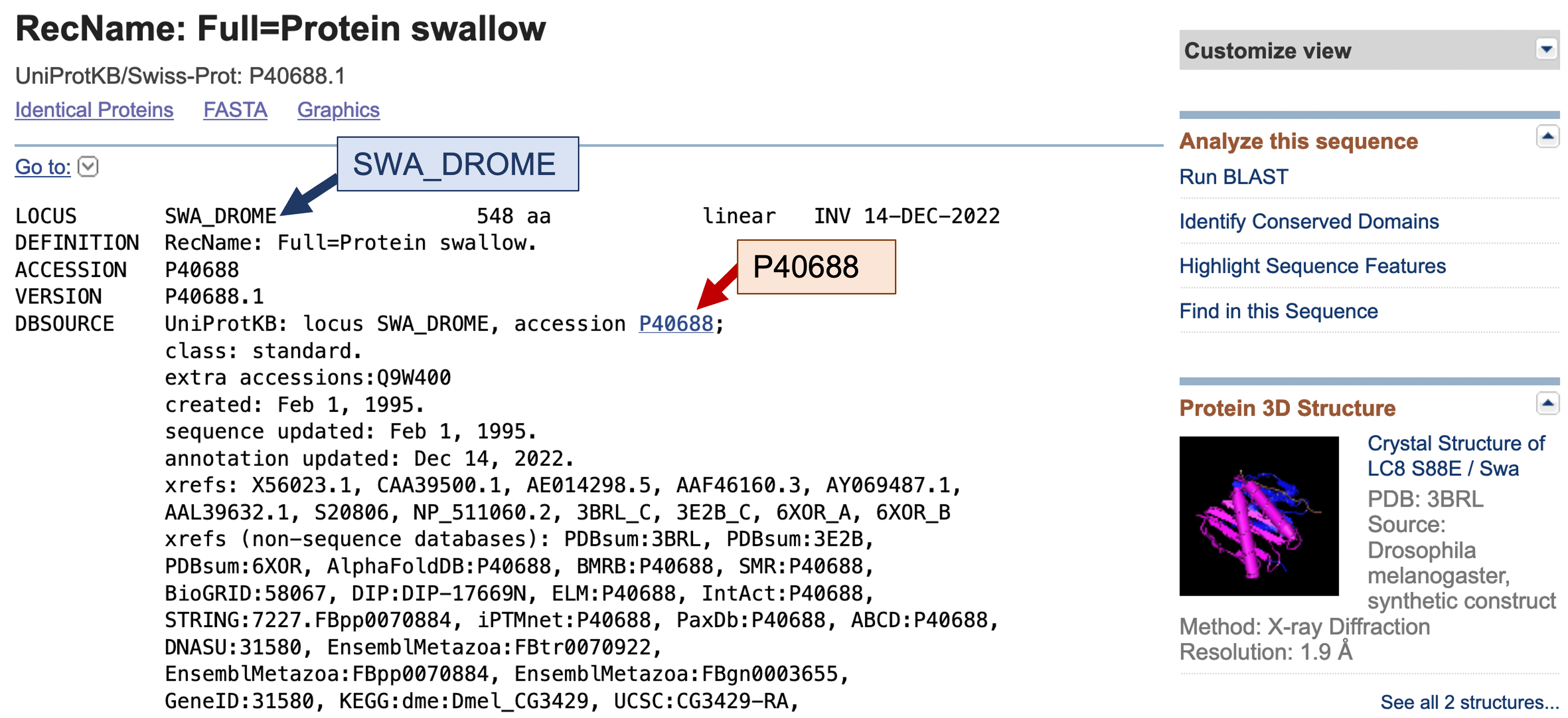
We can navigate to the UniProtKB record for the D. melanogaster swallow protein from the GenBank record directly by clicking on the “P40688” link under the “DBSOURCE” field (red arrow, Figure 9). Alternatively, we can search for the protein record using the accession string “SWA_DROME” at the UniProt website.
|
|
The UniProtKB record for SWA_DROME is also available in the exercise package (file named UniProtEntry.pdf). |
Based on the UniProtKB entry (Figure 10), what does swallow do? Does your blastx output match the swallow genes from more than one species, and if so, which species? If you want to talk about the D. melanogaster and D. pseudoobscura swallow genes in your own work, whom should you cite as having discovered it?
|
|
Check the Publications section of the UniProtKB record. |
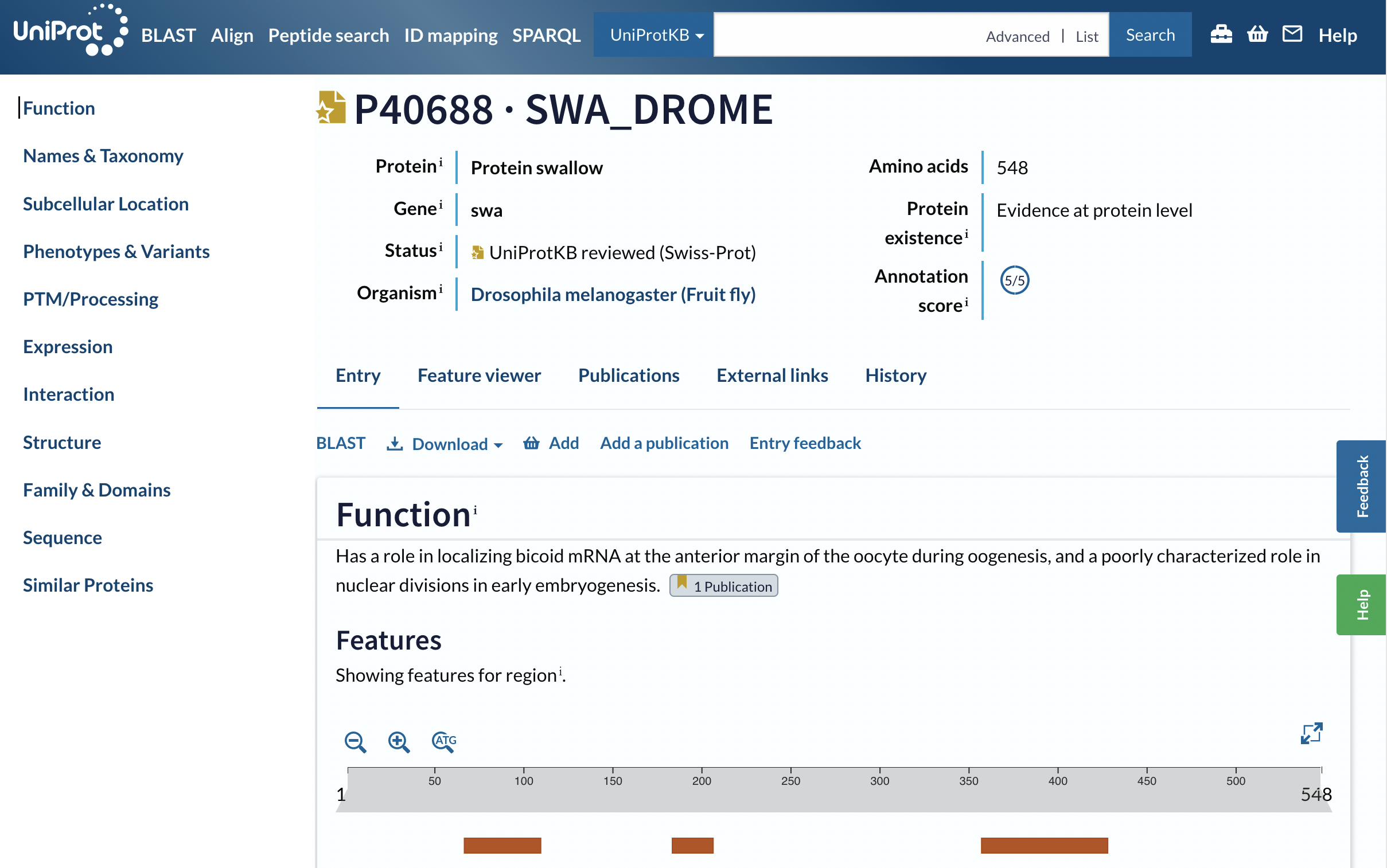
Now that we know a bit more about the candidate matches to our gene, let’s take a closer look at the blastx output. To produce an annotation, we need to verify that the query sequence really does contain the D. melanogaster swallow gene. In particular, the match should be full-length, including all the coding exons of the gene. If there are exons missing, then this may indicate that the feature is a pseudogene.
From the “Graphic Summary” tab of the blastx output, we see that there are many alignment blocks distributed across our entire query sequence (Figure 11). To determine if there are parts of the protein that are missing or if there are multiple hits to the same part of the protein, we need to examine the alignment more closely. Go back to the hit table in the “Descriptions” tab and click on the description for the match with the lowest E-value (i.e., “RecName: Full=Protein swallow [Drosophila melanogaster]” with E-value 2e-174).
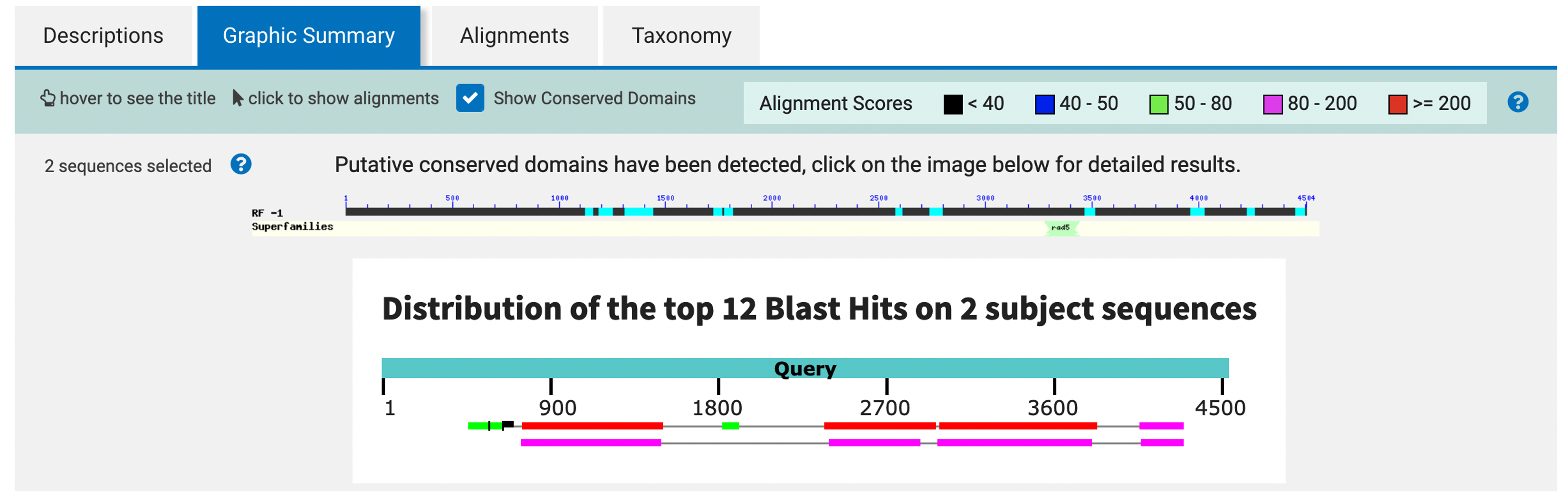
What is the orientation of the swallow gene relative to our query sequence?
|
|
Check the frame in your alignment. |
There is considerable confusion in the collection of blastx alignments to the SWA_DROME protein. As an annotator, your job is to produce order from this chaos.
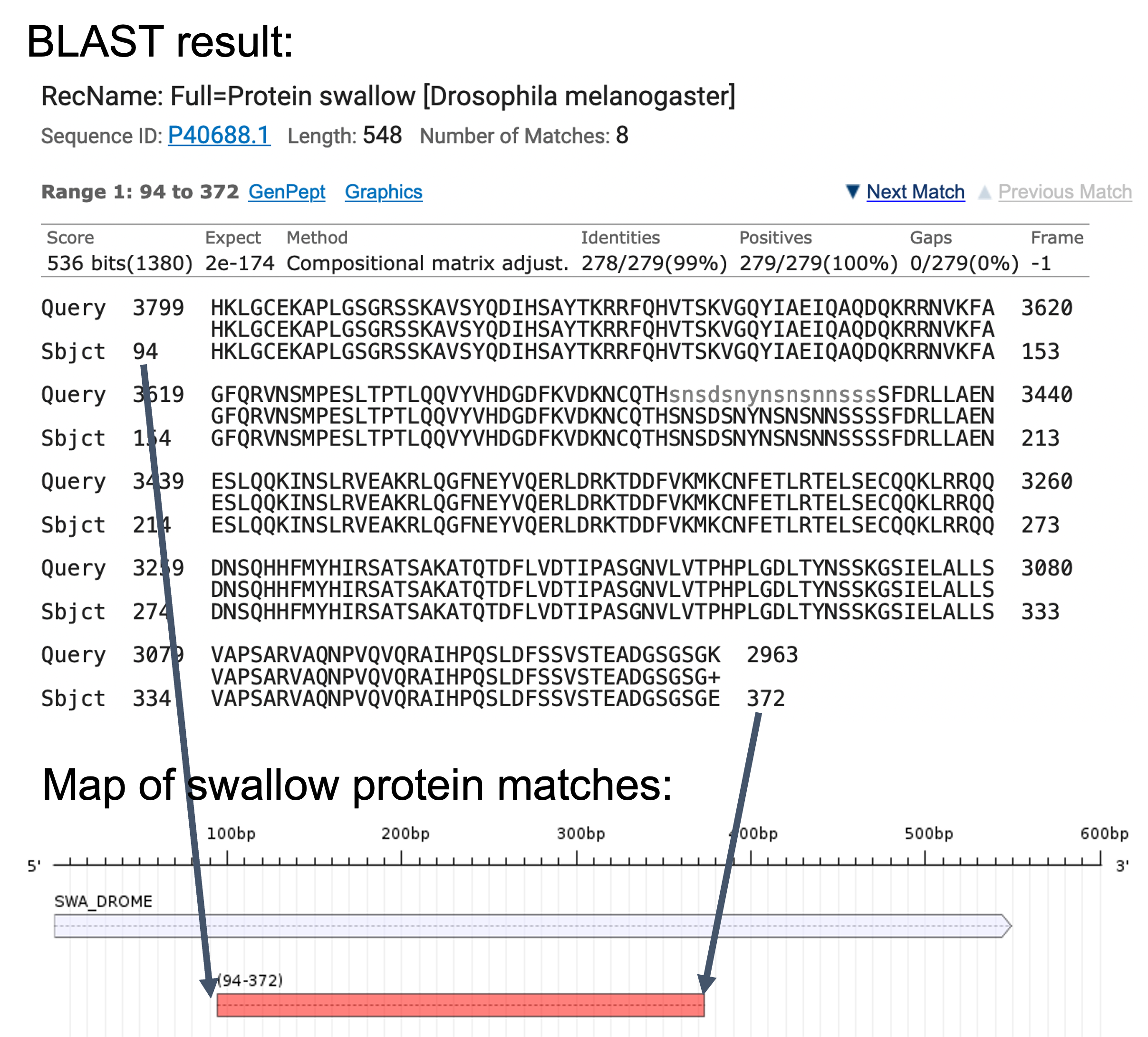
Look at all the matches to SWA_DROME in your blastx output. Is the entire protein matched? If not, which residues are missing? Are there any regions of the protein that are aligned to multiple places in our query sequence?
|
|
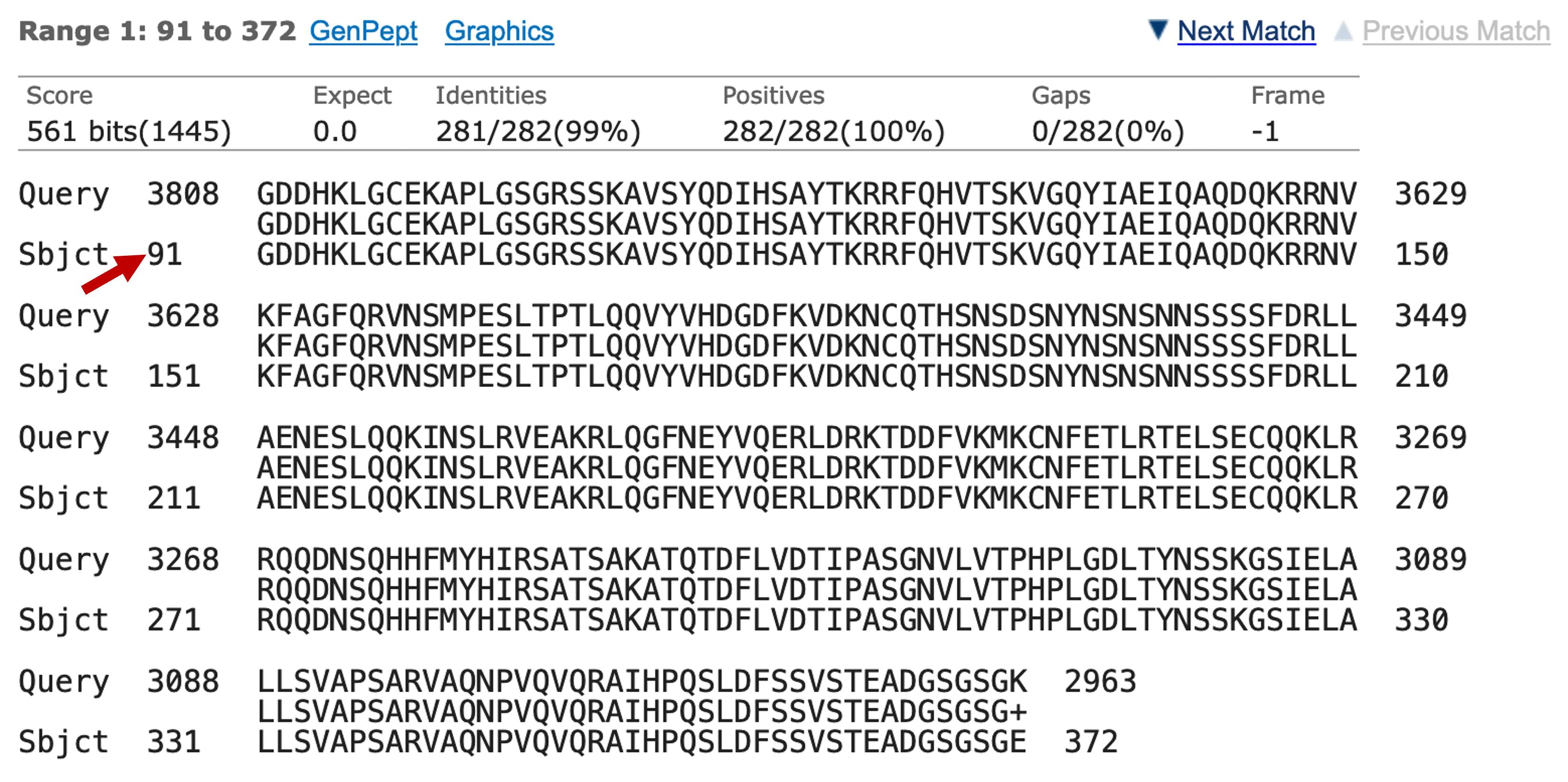
You may find it helpful to draw a figure of the BLAST results in order to check the coordinates of all the alignment blocks relative to the subject (i.e., the swallow protein) sequence. For example, the first alignment block maps to 94-372 of the swallow protein, so on a map we would draw a block that corresponds to this region of the swallow protein (Figure 12). Use the same strategy to draw the rest of the alignment blocks onto the map. |
We will continue our investigation with a more detail analysis of the missing residues. Go back to the UniProtKB entry for SWA_DROME and find the part of the protein that is not represented by any of the BLAST alignments between SWA_DROME and our sequence, as shown by your analysis in Question 8.
Which amino acids predominate in the missing region? Given that blastx will, by default, mask low-complexity sequence in the query before a search, do you have a reasonable explanation for why this part of the protein is missing? What evidence would support your hypothesis?
Around amino acid 190 of the protein (subject) sequence, you will see a series of gray letters representing masked residues (Figure 13) in the query. BLAST apparently decided that the protein in the region, rich in serines and asparagines, should be marked as low-complexity. Aligning a residue to a masked base yields a negative score.
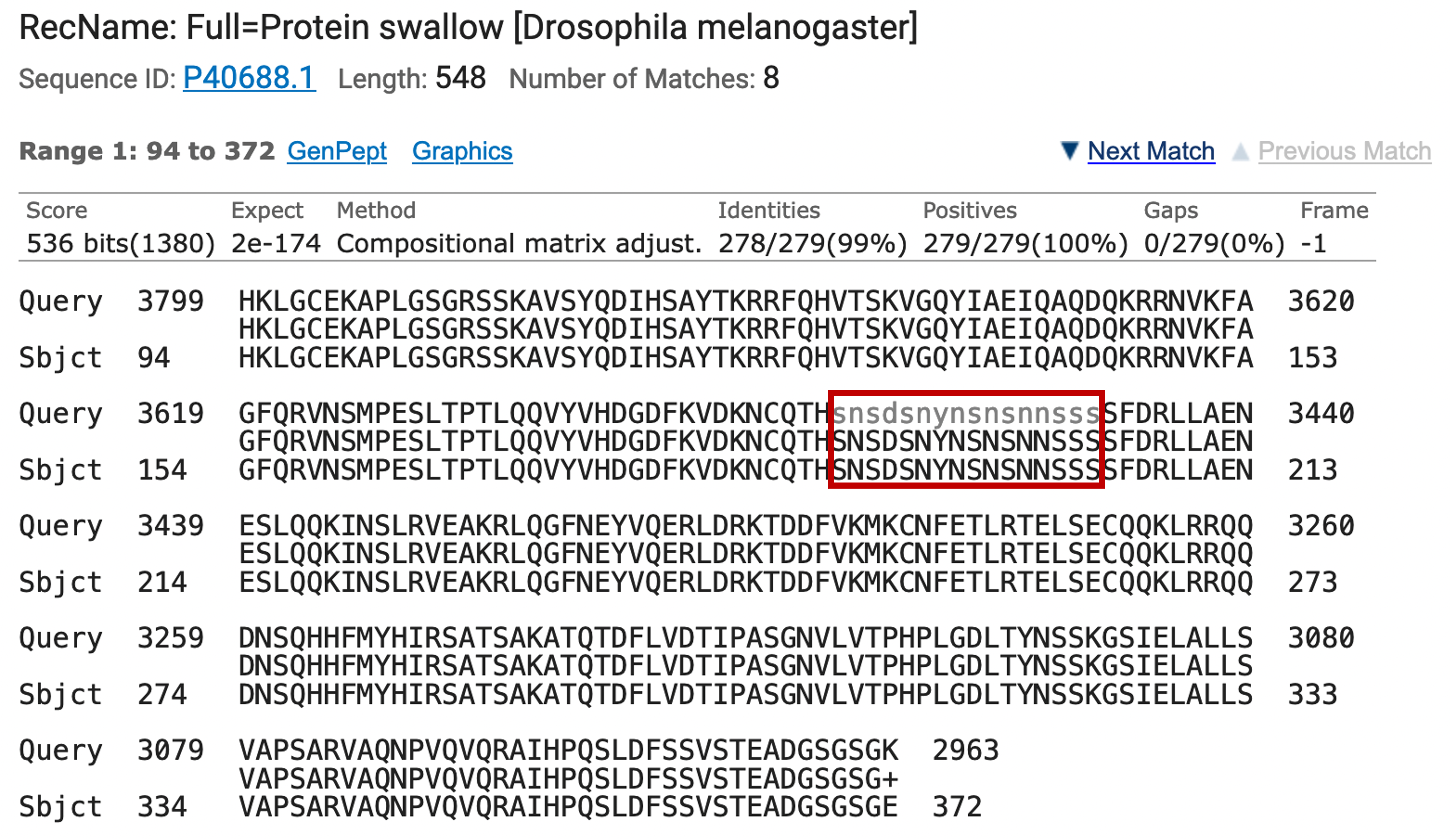
Given blastx seems happy enough to include masked residues in its alignments (as shown in the figure above), why didn’t it include residues 78-93 of the protein?
|
|
Look at the reading frames (specified in the header) of the matches ending at 77 and beginning at 94. What happens if you add negative-scoring residue pairs to the end of an alignment? |
With careful inspection of the results, you should see that most of the residues of the swallow protein align to two places in the query. We will need to investigate this further in order to fully annotate the query. To get a more detailed graphical representation of all the alignment blocks between the swallow protein and the query sequences, we will go back and perform another blastx search. Start by clicking on the “Edit Search” button at the top of the blastx output to go back to the BLAST input page. Because we are interested specifically in the swallow protein, we will use BLAST to directly align this protein sequence with our query.
Click on the “Align two or more sequences” checkbox on the BLAST input page. This will change the BLAST input page so that the “Choose Search Set” section is replaced by the “Enter Subject Sequence” section. As before, click on the “Browse” or “Choose File” button under the “Enter Query Sequence” section and then select the dmel_seq2.fasta file from the exercise package. In the bottom text box, we want to enter the amino acid sequence of the swallow protein. We could retrieve the sequence from the GenBank record, copy the sequence, and then paste it into the text box. However, BLAST will automatically retrieve the protein sequence if we enter the accession number of the swallow protein into the text box. Looking back at the GenBank record for our first result (Figure 8), we see that the accession number for the swallow protein is P40688. Enter “P40688” into the “Enter Subject Sequence” text box to indicate that you want to use the swallow protein as the subject sequence in your blastx search (Figure 14).
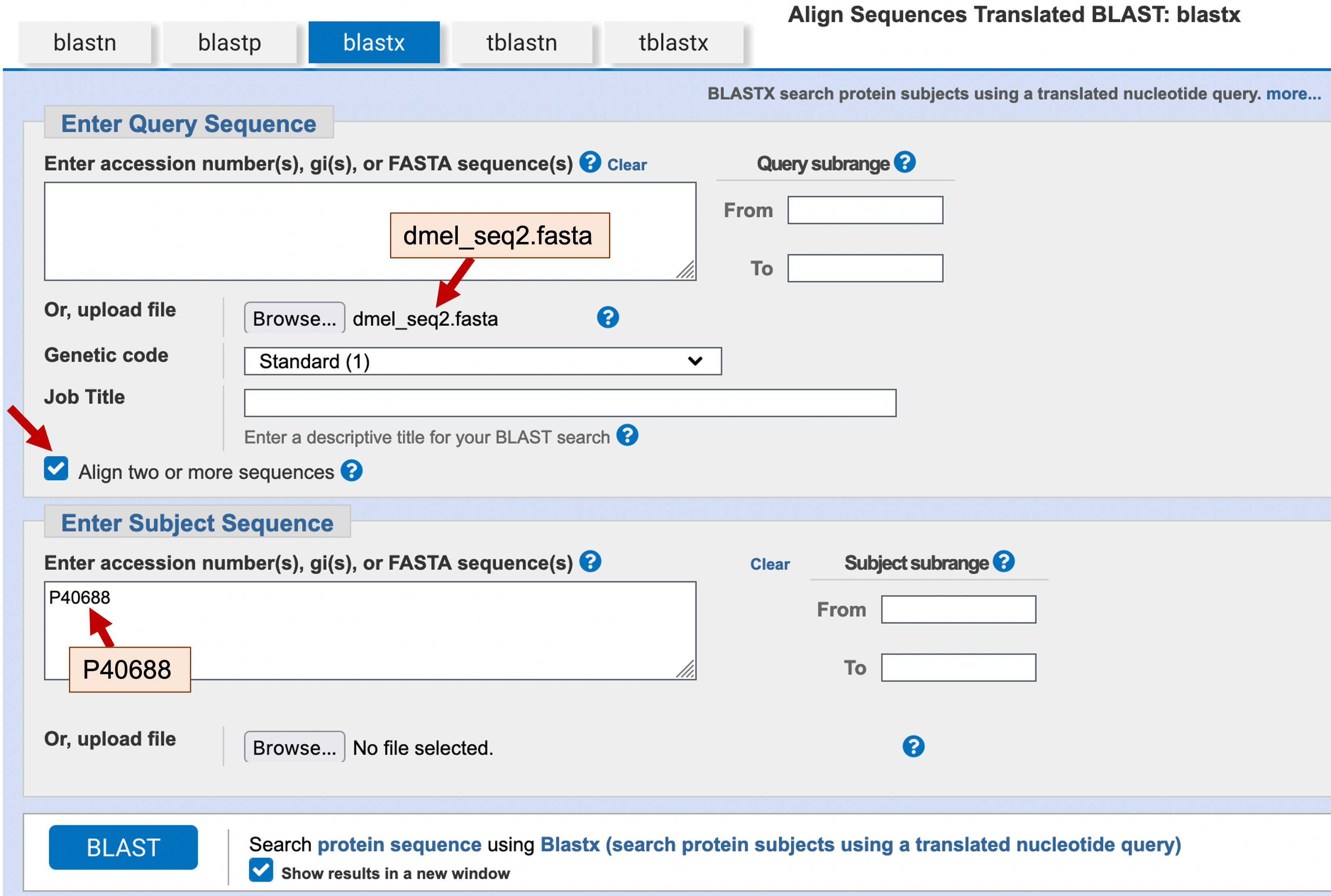
To determine if the low complexity filter is the cause of the missing residues in our original blastx alignment, we will turn off the low complexity filter when we run this BLAST search. Click on the “Algorithm parameters” header to expand this section and then uncheck the option “Low complexity regions” under the “Filters and Masking” section. In addition, we will also change the “Compositional adjustments” field to “No adjustment”. The use of a compositionally adjusted scoring matrix will generally produce BLAST results with more accurate E-values and reduce the number of spurious matches when searching a large database. However, this adjustment could remove conserved residues from the alignment when we are comparing only two sequences against each other.
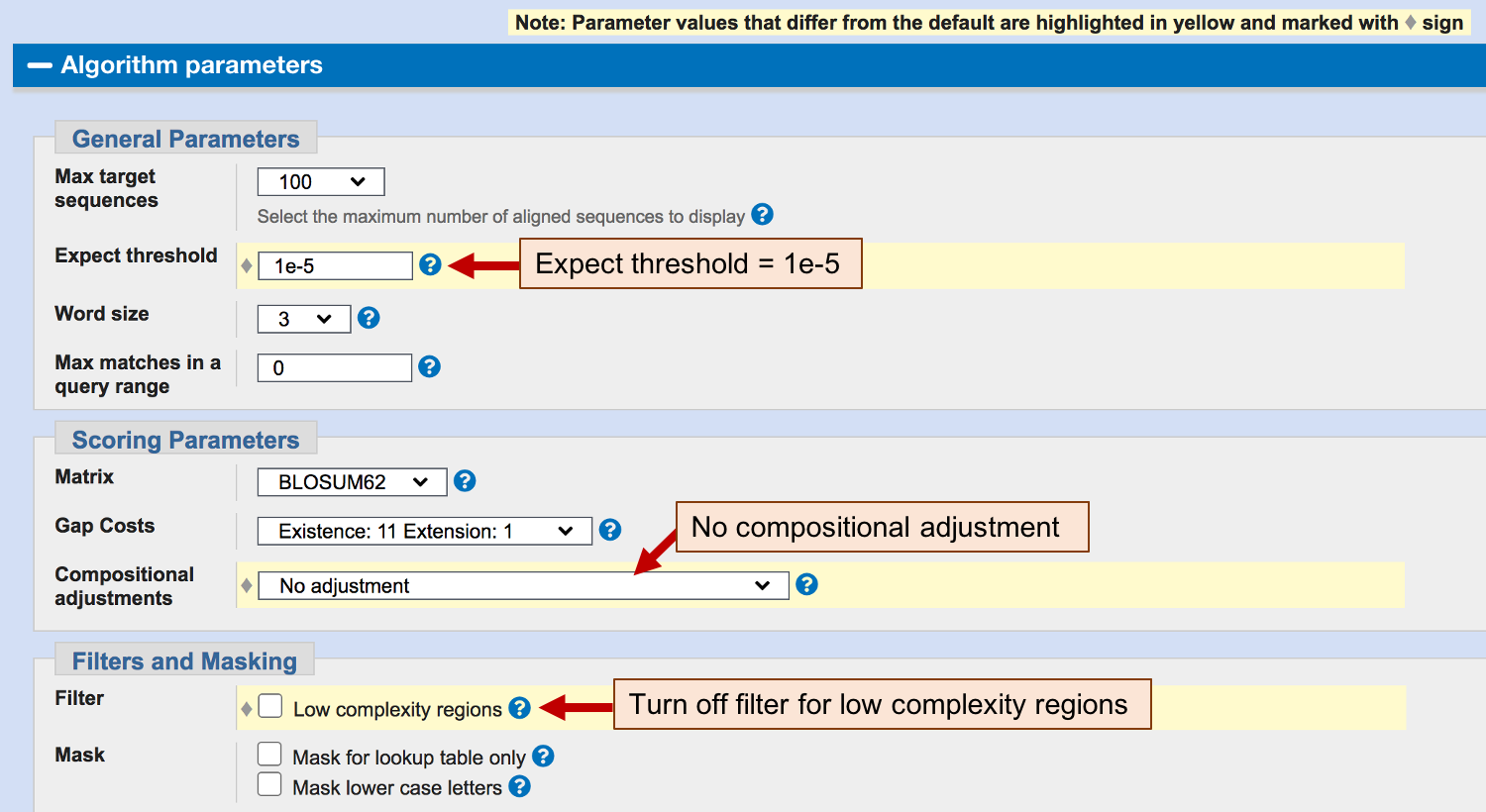
Because our previous BLAST results show the relevant alignments all have E-values below 1e-5, we will also set the “Expect threshold” to “1e-5” (Figure 15). In addition, verify that the “Word size” parameter is set to 3. Click on the “BLAST” button after you have changed these settings.
|
|
For teaching purposes, the blastx comparison of these two sequences is available in the exercise package (blx_swallow_nolow_dmel_seq2.txt). |
Click on the “Alignments” tab to examine the blastx alignment. We found that the original alignment block that spans from 1-77 of the swallow protein has been extended to 1-91 in the new blastx result (Figure 16).
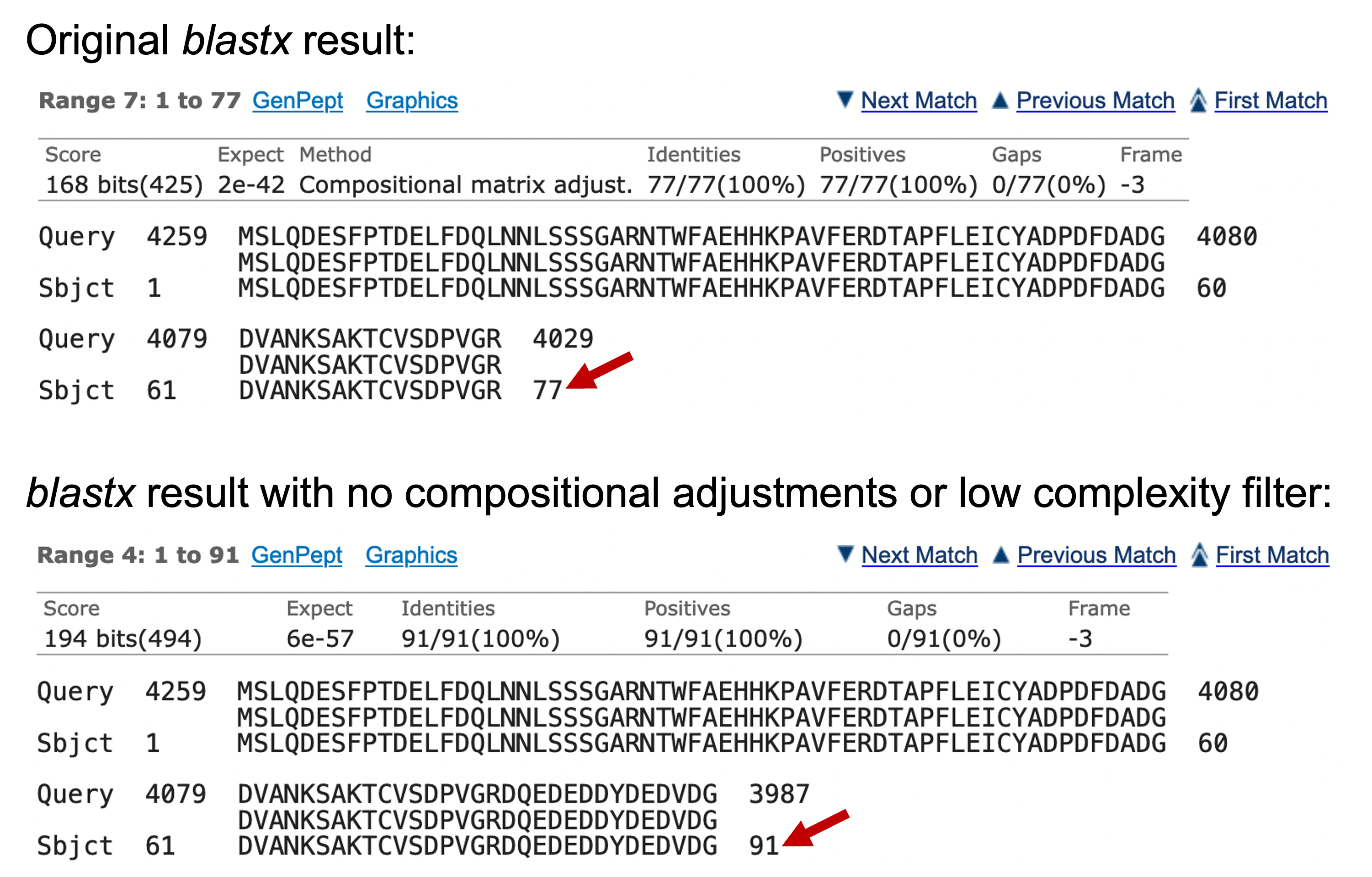
Similarly, the first alignment block in the original blastx search result covers residues 94-372 of the swallow protein. After turning off the filters, the beginning of the blastx alignment block has been extended to cover residues 91-372 of the swallow protein (Figure 17).
Now that we have accounted for all the missing amino acids in the swallow protein, we should get an overall view of how all the alignment blocks are organized. Click on the “Dot Plot” tab in the blastx output. This will open a section that contains a dot matrix comparison of the query and subject sequences. The x-axis of the dot matrix corresponds to the query sequence and the y-axis corresponds to the swallow protein sequence. The lines within the dot matrix correspond to the positions of the alignment blocks.
Looking at these results, how many “swallow” like features are found in the query, which (if any) seems most likely to be the true swallow gene? What might the other matches be, and what biological mechanisms might have produced them?
Further exploration Using blastn
To make further progress in determining the correct annotation for our sequence, we will try to obtain additional evidence at the nucleotide level, specifically looking at Drosophila mRNAs.
In order to detect homology at the nucleotide level, we will use nucleotide BLAST (blastn) instead of blastx. As was the case for the blastx search, we must make some decisions before we can perform the search. For example, we could compare our query sequence against the Core nucleotide database (core_nt) or one of the EST databases. Because ESTs are pretty noisy and do not come with easily accessible annotations, we will use the core_nt database in this exercise.
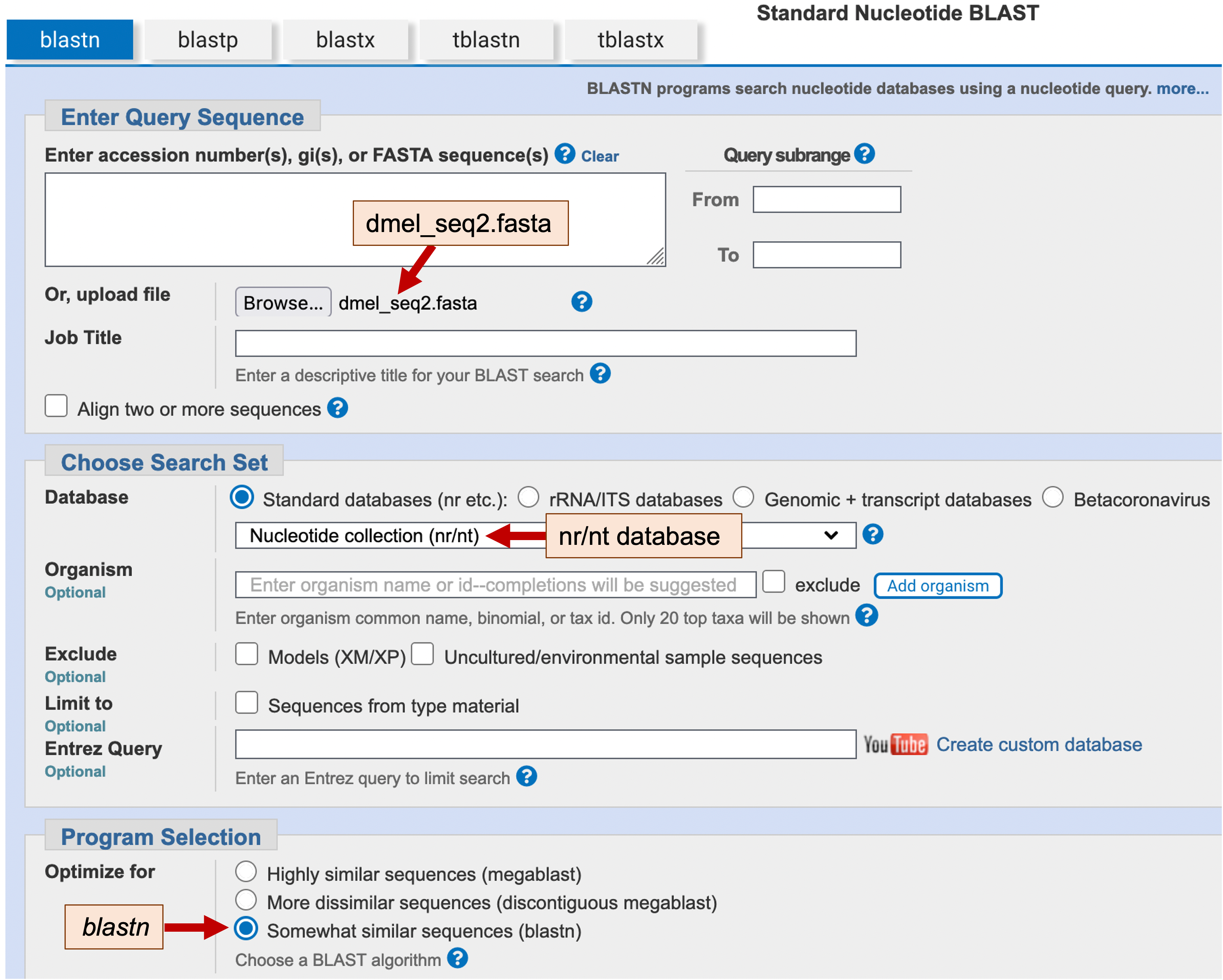
To do the blastn search, go back to the BLAST page and click on the “Nucleotide BLAST” image under the “Web BLAST” section. As before, click on the “Browse” or “Choose File” button under the “Enter Query Sequence” section, and then select the dmel_seq2.fasta file from the exercise package. Select “Core nucleotide database (core_nt)” in the “Database” field. Lastly, because blastn is more sensitive than megablast, we will select “Somewhat similar sequences (blastn)” under the “Program Selection” field (Figure 18). Click on the “BLAST” button and then wait for your results.
|
|
Alternatively, the blastn output is available in the exercise package (bln_core_nt_dmel_seq2.txt). |
Had your query contained a repetitive element such as a transposon, what would have happened had you forgotten to repeat-mask the query sequence before running the BLAST search?
Sequences in the GenBank Nucleotide collection (nr/nt) database come from many sources, including genomic contigs from whole-genome sequencing projects and mRNAs/cDNAs. The core_nt database is a smaller database which contains all the transcript and gene-related sequences in the nr/nt database. A particular useful class of mRNA entries is the NCBI RefSeqs, these sequences come from a curated database of full-length mRNAs from various genes. You can find out more information about the RefSeq database through the “Using RefSeq” section of the RefSeq home page. You can easily recognize RefSeq matches in the BLAST output because their accession numbers always begin with two letters (NM or XM for mRNA records and NP or XP for protein records), followed by an underscore and a unique ID (Figure 19). See Table 1 in Chapter 18 of the NCBI Handbook for the list of accession prefixes used by the RefSeq database.
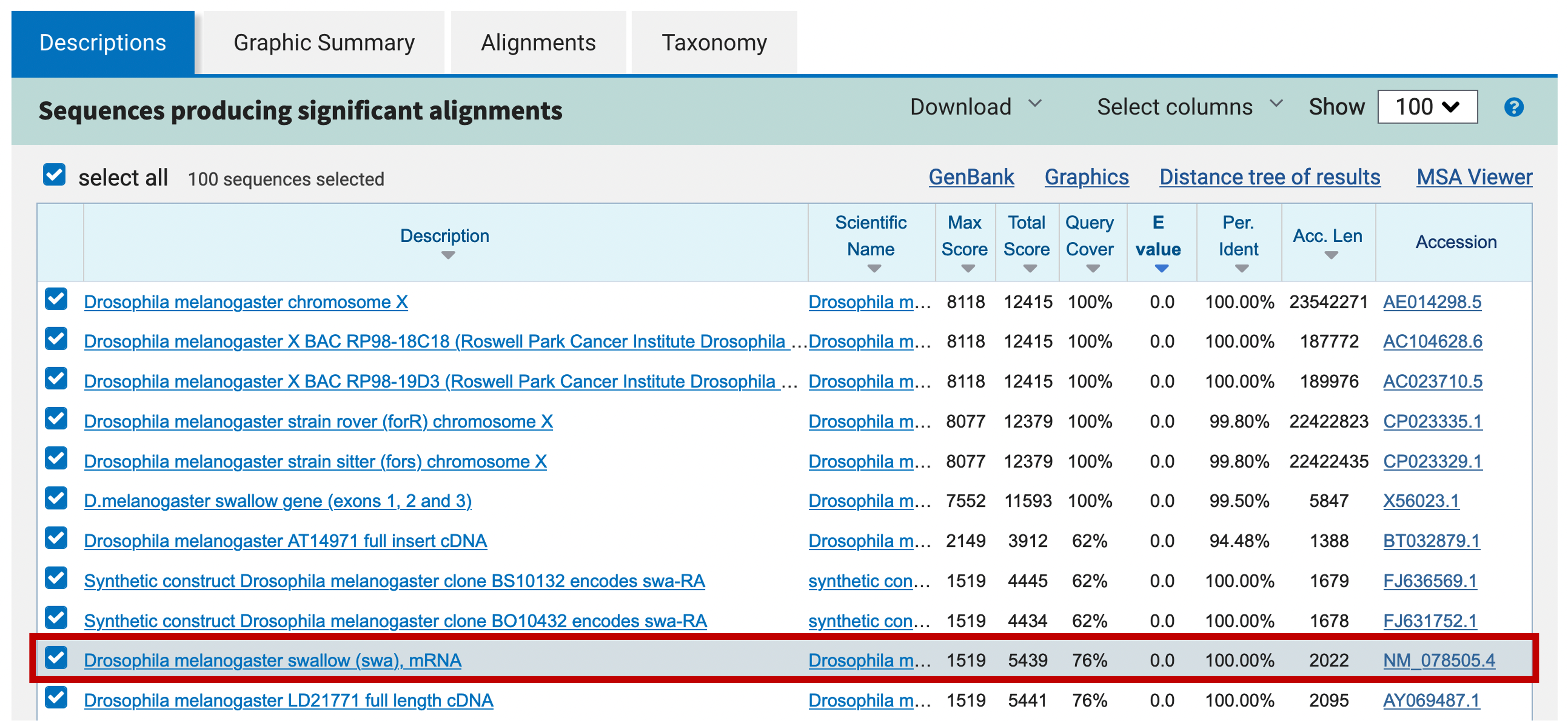
Click on the description for the D. melanogaster swallow RefSeq mRNA (NM_078505.4) in order to navigate to the corresponding blastn alignment.
What is the best RefSeq match to the query? How good is the match to what you think is the true swallow gene? Based on the blastn alignment, how many exons does the gene have, and roughly where do the introns occur?
|
|
Use Figure 20 below to create a map showing all the blastn hits to the query sequence as you did for Question 8. |
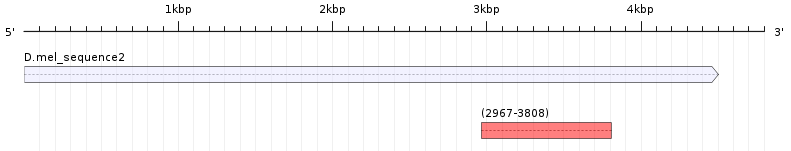
How well does the RefSeq match the other part of the query? Can you see regions that were not aligned at the protein level? Why might this be?
The main question at this point is what is the other set of matches outside the region that we believe to be the D. melanogaster swallow gene? Do these matches reveal a real gene or pseudogene? Pseudogenes are rare in Drosophila compared to mammals, but they are not unknown.
There are two signals that strongly suggest a putative match to a gene might be a pseudogene: internal stop codons that produce a truncated protein and gaps in the alignment that causes frame shift mutations.
The blastx alignment uses an asterisk (*) to represent a stop codon in the alignment.
Gaps in the coding region of a blastn alignment would result in a frame-shift if the size of the gap were indivisible by three.
Frame shift mutations may introduce internal stop codons that prematurely terminate the translation of the protein.
Keeping in mind the exon boundaries you inferred above, could you find evidence of premature stop codons and/or frame shift-inducing gaps that would cause you to diagnose a pseudogene adjacent to the swallow gene? Describe any evidence you find.
Summary
Based on all the evidence gathered in this exercise, how would you annotate the query sequence? What uncertainties remain? Compose a short (a few sentences) paragraph that you could add to an annotation database summarizing your findings.