Resources & Tools
-
The package containing the files for this exercise are available through the “An Introduction to NCBI BLAST” page on the GEP website.
Introduction
The Basic Local Alignment Search Tool (BLAST) is a program that can detect sequence similarity between a query sequence and sequences within a database. The ability to detect sequence similarity allows us to identify putative orthologous genes in a sequence from another species. It also allows us to determine if a gene or a protein is related to other known genes or proteins.
BLAST is popular because it can quickly identify regions of local similarity between two sequences, which we can then use to infer homologous sequences. More importantly, BLAST uses a robust statistical framework that can determine if the alignment between two sequences is statistically significant. In this exercise, we will use the National Center for Biotechnology Information (NCBI) BLAST service to help us annotate a sequence from the Drosophila yakuba genome (unknown.fna in the exercise package).
The NCBI BLAST web interface
Before we begin the analysis, we should first familiarize ourselves with the NCBI BLAST web interface. Open a new web browser window and navigate to the NCBI BLAST home page. In this exercise, we will only use a few of the tools available on the NCBI BLAST website. To learn about the more advanced options available (such as setting up My NCBI accounts), click on the “Help” link on the main navigation bar to access the documentations for NCBI BLAST (Figure 1).
All the NCBI BLAST pages have the same header with four links:
| Links | Explanation |
|---|---|
Home |
Link to the NCBI BLAST home page |
Recent Results |
Link to results of the BLAST searches you have run previously |
Saved Strategies |
NCBI BLAST search parameters you have previously saved to your My NCBI account |
Help |
Documentation for NCBI BLAST |
Besides the main toolbar, there are two other sections of the NCBI BLAST web interface that are of interests: the “Web BLAST” section contains links to the common BLAST programs and the “Specialized searches” section contains links to additional tools for performing sequence searches (e.g., use CD-search to identify conserved domains within a query sequence). The type of BLAST search you need to use will depend primarily on the type of query sequence and the database you would like to search.
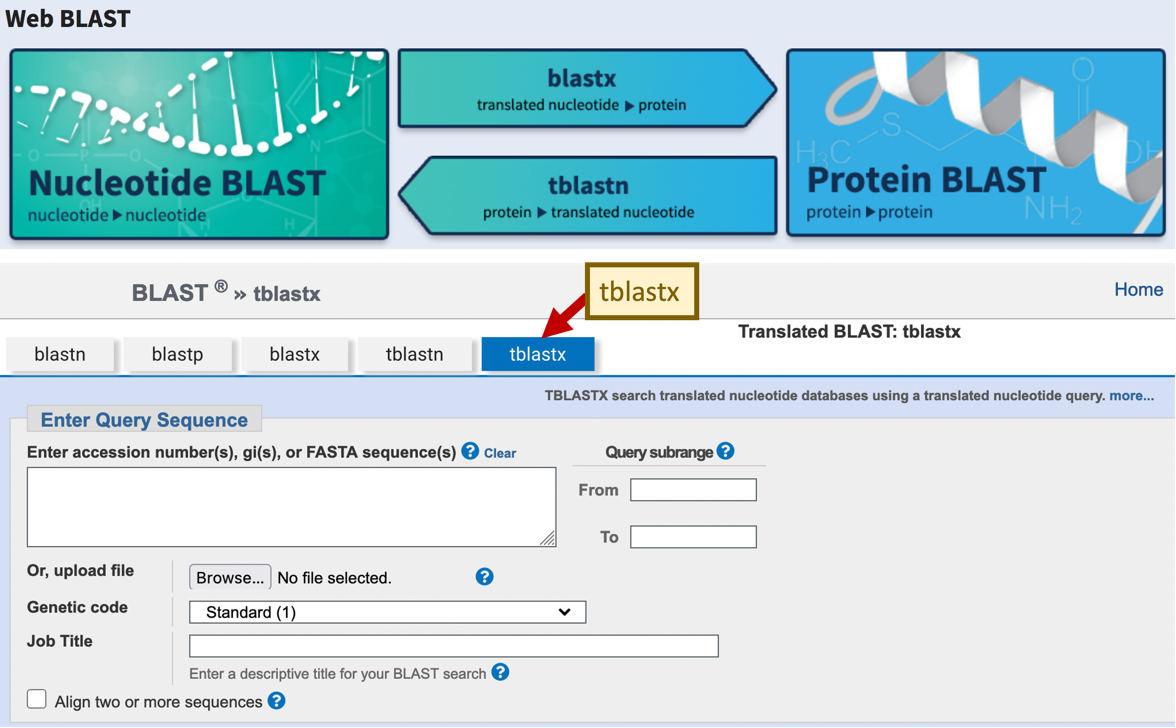
Four of the five common BLAST programs are available through the “Web BLAST” section of the NCBI BLAST home page (Figure 2, top). The program tblastx, which translates the nucleotide query and nucleotide database when it performs the sequence comparisons, is not listed under the “Web BLAST” section. However, you can access this program by clicking on any of the BLAST programs in the “Web BLAST” section and then click on the “tblastx” tab in the NCBI BLAST search form (Figure 2, bottom).
|
|
The tblastx program can be used to identify protein-coding genes in two unannotated genomes. This strategy could be used to identify novel genes in the target species that are absent from the informant species (e.g., D. melanogaster) but is present in a closely-related species. Some gene prediction algorithms (such as the SGP2 gene predictor used by the GEP annotation projects) incorporate tblastx alignments as an additional line of evidence to improve the accuracy of gene predictions. |
The basic BLAST programs are summarized below:
| BLAST program | Query | Database |
|---|---|---|
Nucleotide BLAST (blastn) |
Nucleotide |
Nucleotide |
Protein BLAST (blastp) |
Protein |
Protein |
blastx |
Translated Nucleotide |
Protein |
tblastn |
Protein |
Translated Nucleotide |
tblastx |
Translated Nucleotide |
Translated Nucleotide |
Finding similar nucleotide sequences using blastn
In this exercise, we will characterize an unknown genomic sequence (unknown.fna) and determine if it has sequence similarity to any known genes. One strategy we can use is to search for sequence similarity to mRNA sequences in the NCBI Reference Sequence (RefSeq) database.
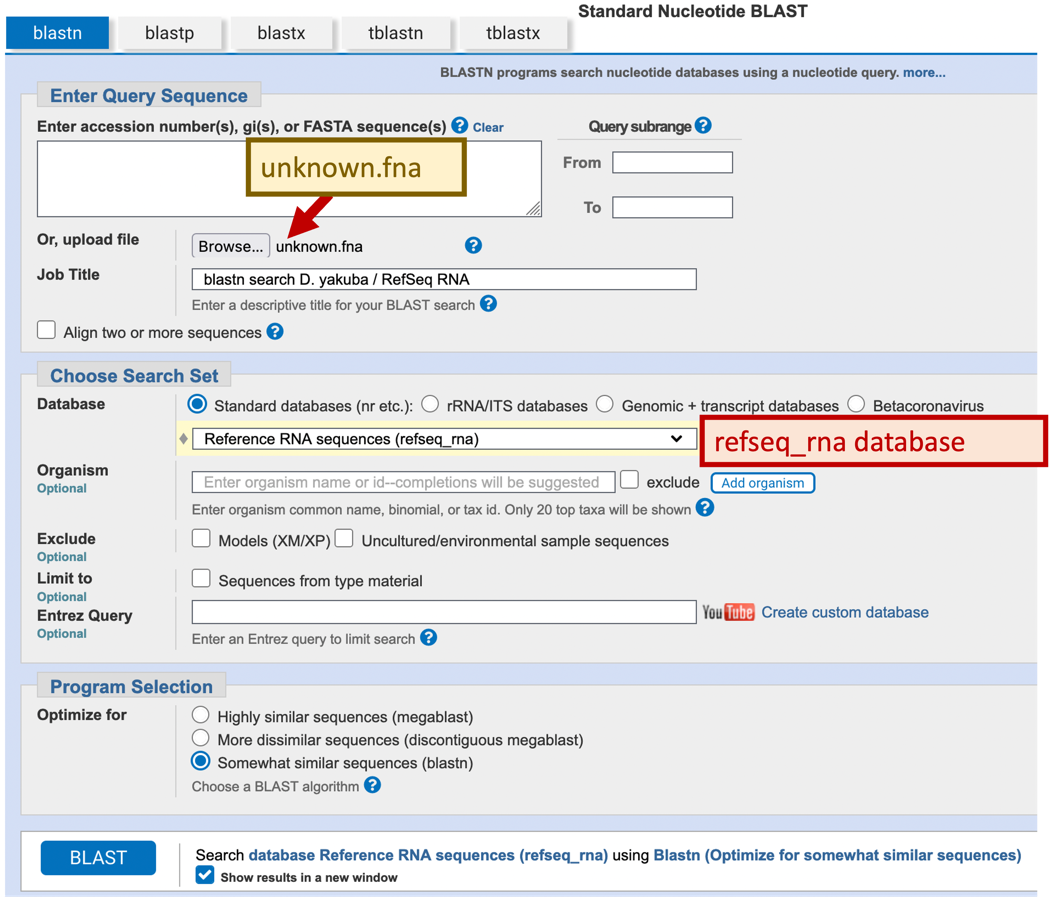
When we set up a BLAST search, there are three basic decisions we must make: the BLAST program we want to use, the query sequence we want to annotate, and the database we want to search. In addition, we can change several optional parameters (such as the Expect threshold and low complexity filters) in order to modify the behavior of BLAST. In this case, we will set up our BLAST search using mostly default parameters. We will use the blastn program to search our sequence (query) against the NCBI Reference Sequence (RefSeq) RNA database (Figure 3).
-
Navigate to the NCBI BLAST home page and click on the “Nucleotide BLAST” image under the “Web BLAST” section
-
Under the “Enter Query Sequence” section, click on the “Browse” or the “Choose File” button and select the file with the unknown sequence (unknown.fna)
-
Enter the Job Title “blastn search D. yakuba / RefSeq RNA”
-
In the “Choose Search Set” section, change the database to “Reference RNA sequences (refseq_rna)”
-
Under “Program Selection”, select “Somewhat similar sequences (blastn)”
-
Check the box “Show results in a new window” next to the “BLAST” button
-
Click “BLAST”
|
|
the blastn search may take a few minutes to complete if the NCBI web server is busy (Figure 4). |
Once the search is complete, a new web page will appear with the BLAST report. For teaching purposes, the BLAST output (blastnInitial.txt) is available in the package for this exercise.
The top left panel (Figure 5) of the BLAST results page shows the parameters used in the BLAST search (e.g., database name, query ID, query length). The controls in the top right panel (Figure 5) can be used to filter the BLAST hits by organism, percent identity, and Expect value (E-value).
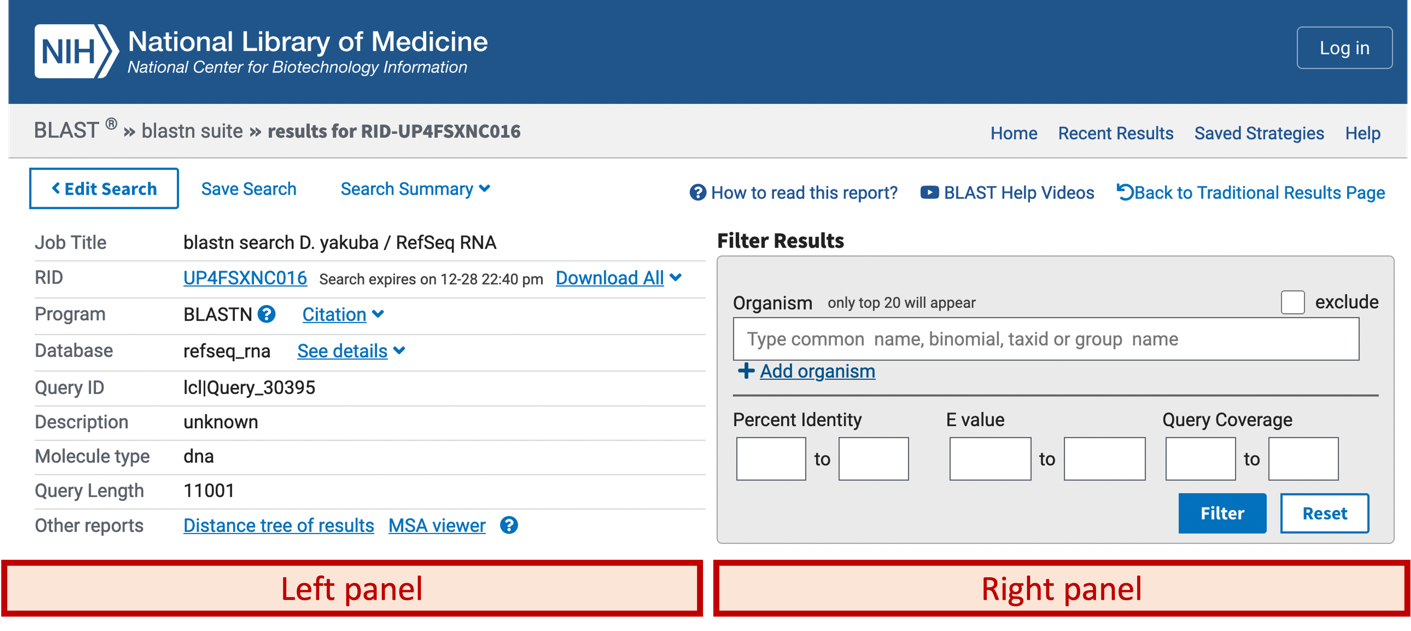
The details of the BLAST results are organized into the four tabs below these two panels: “Descriptions”, “Graphic Summary”, “Alignments”, and “Taxonomy”. We will go through each of these sections in order to interpret our blastn output.
I. Descriptions
This tab shows the list of sequences in the database where the E-value for the alignment to the query sequence is less than or equal to the Expect threshold (e.g., the default Expect threshold is 0.05) (Figure 6). By default, the results are sorted by their E-value in ascending order, where alignments with lower E-values are less likely to occur by chance than alignments with higher E-values. You can click on the column headers to sort the results by the other columns. You can also use the “Select columns” drop-down menu on the main toolbar to show or hide each column.
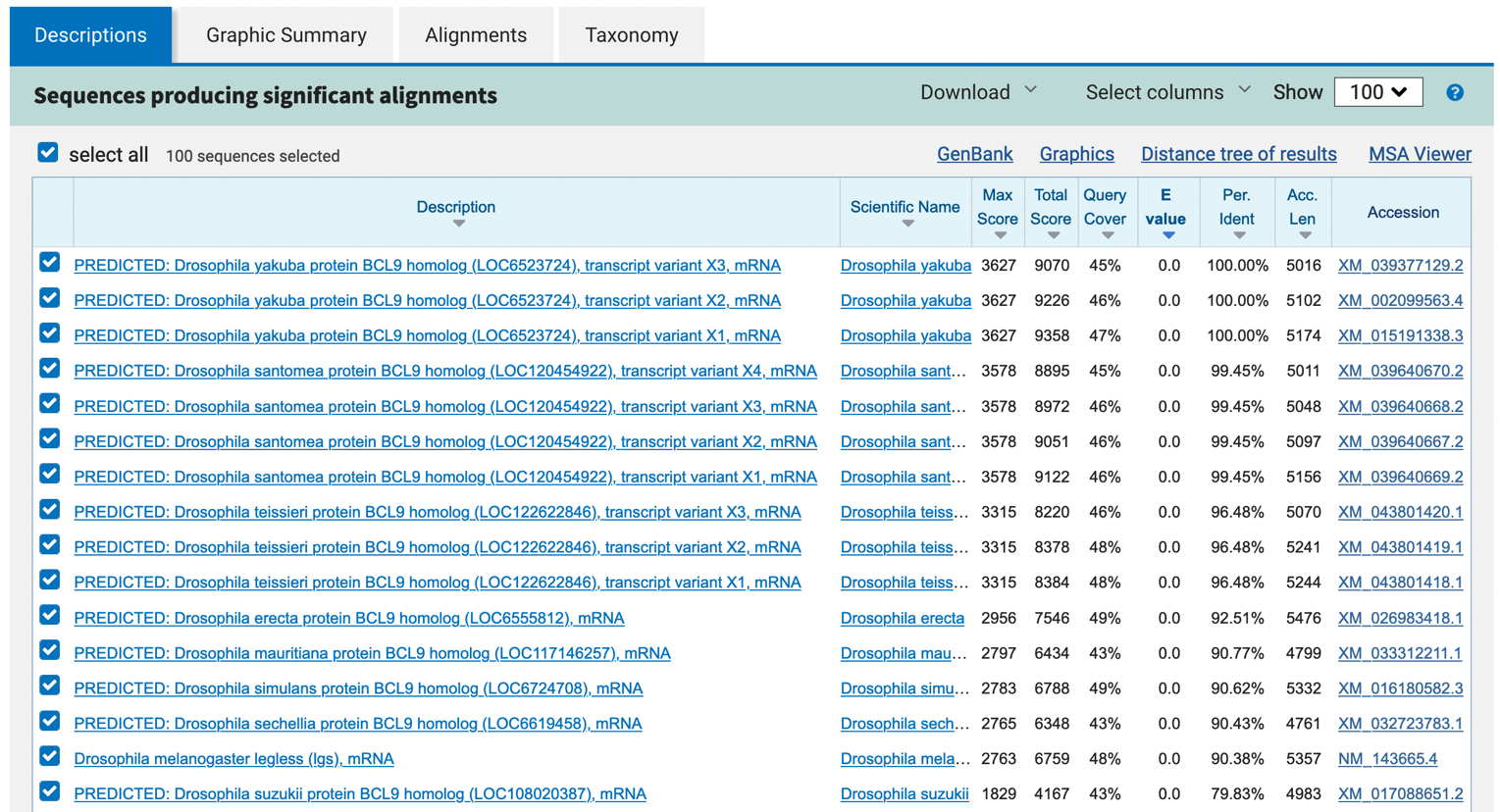
Clicking on the accession number in the table will bring up a new page with the GenBank record of the sequence. Clicking on the description of the hit will bring us to the corresponding alignment in the BLAST output. Alternatively, you can click on the “Alignments” tab to jump to the first alignment.
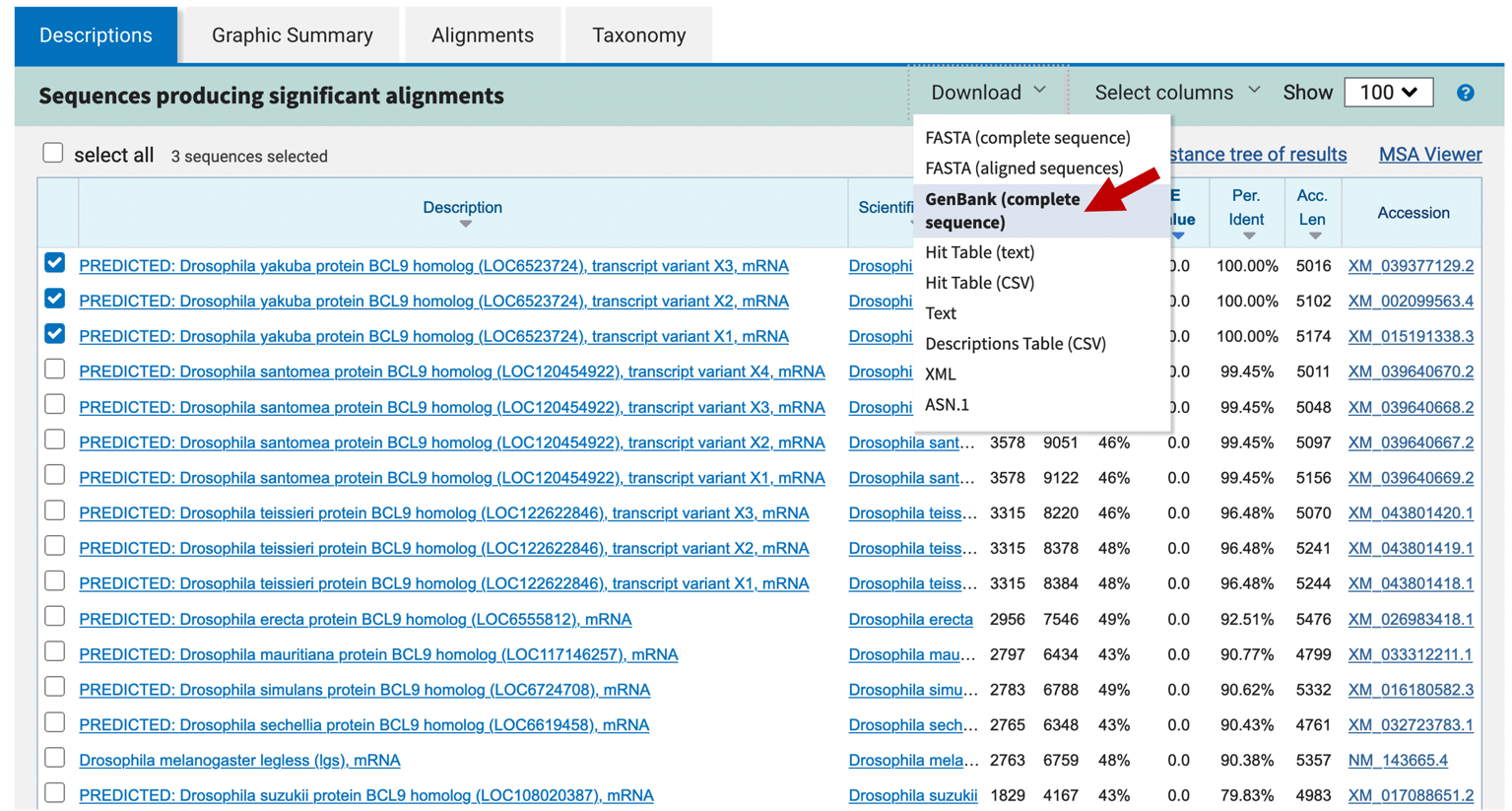
In addition to reviewing the records for individual sequences, you can also review multiple sequence records by selecting the checkbox next to each match. The contents of the other tabs will update automatically based on your selection. You can use the “Download” drop-down menu on the main toolbar to download the selected hits in multiple formats (e.g., FASTA, GenBank, Hit Table). For example, we can use the following steps to retrieve the GenBank records for the first three D. yakuba blastn hits in the Descriptions table (Figure 7):
-
Uncheck the “select all” checkbox above the BLAST hit table
-
Select the checkboxes for the first three blastn hits
-
Click on the “Download” drop-down menu on the main toolbar, and then select the “GenBank (completesequence)” [sic] option
II. Graphic Summary
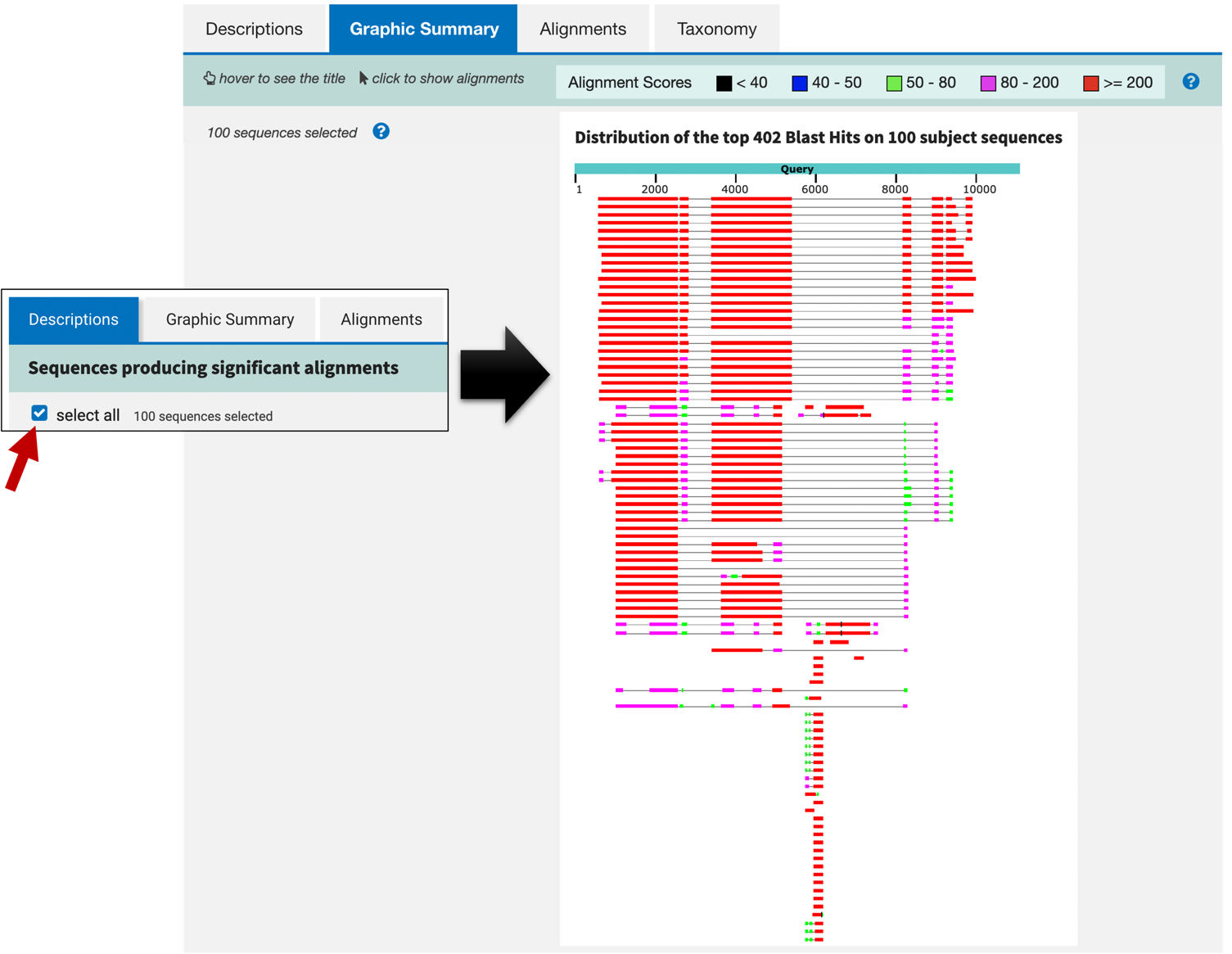
This tab provides a graphical overview of the alignments between the selected BLAST hits in the Descriptions tab and the query sequence. The boxes correspond to regions in the query that have sequence similarity to the sequences in the database. The color of the box corresponds to the score, where hits with higher scores are more significant. When you move your mouse over a BLAST hit, the title of the subject sequence will appear in a tooltip. Click on the color box and then click on the “Alignment” link to jump to the alignments associated with that BLAST hit.
To examine the graphical overview for all the blastn hits, go back to the “Descriptions” tab and then select the “select all” checkbox. Click on the “Graphic Summary” tab to view the updated graphical overview (Figure 8).
III. Alignments
This tab contains the alignments between the selected BLAST hits in the Descriptions tab and the query sequence. The sequence alignments show us how well the query sequence match the subject sequence in the database. Because we will rely on sequence alignments heavily in our annotation efforts, we will examine this Alignment tab more closely.
Alignments to different subject sequences in the database are separated by a blue toolbar that contains options to manipulate the alignment results and to retrieve additional information for that specific BLAST hit (Figure 9). For example, we can use the “Download” drop-down menu on this toolbar to obtain the FASTA sequence or the GenBank record for a specific hit. We can use the navigation links at the right side of the toolbar to quickly navigate to the next or the previous BLAST hit.
In addition, we can click on the “Graphics” link to examine the location of each alignment block relative to the subject sequence (Figure 10).
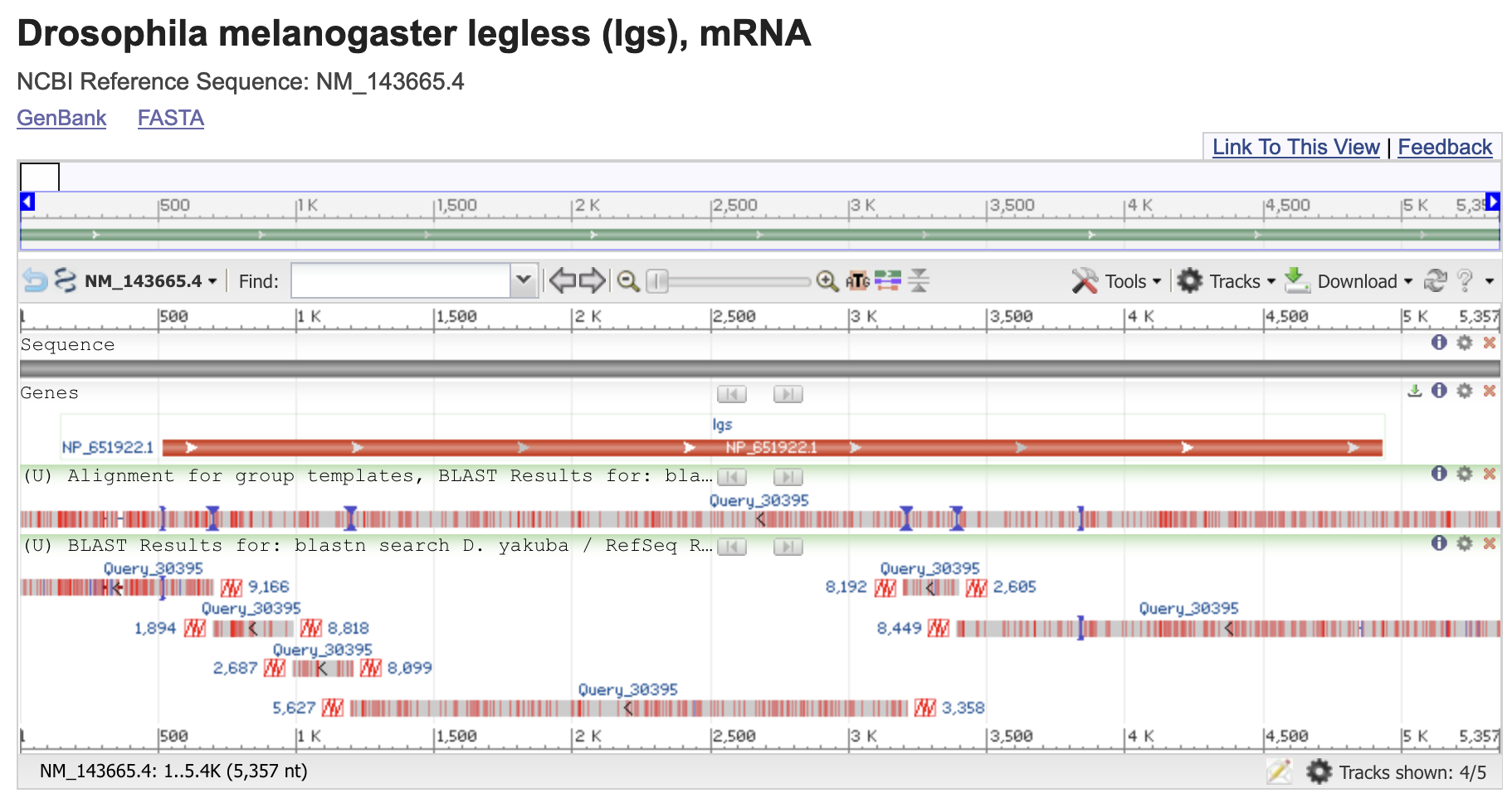
As its name suggests, BLAST is designed to identify local regions of sequence similarity. This means that BLAST might report multiple distinct regions of sequence similarity when we align a query against a subject sequence in a database. For example, if we were to align a processed mRNA sequence to a genomic sequence, we would expect to see multiple alignment blocks (many of which correspond to transcribed exons) in our BLAST output. Each alignment block demarcates a local region of similarity between the query and the subject sequences. Regions of the genomic sequence without significant alignments that fall between these alignment blocks would likely correspond to intronic sequences.
The “Number of Matches” field beneath the name of the sequence shows the number of alignment blocks identified by blastn. For example, the blastn hit for the legless mRNA from D. melanogaster contains six different alignment blocks to the subject sequence — (Figure 11). Each alignment block represents a region of the D. melanogaster legless gene that shows sequence similarity with our genomic sequence from D. yakuba.
You can use the “Sort by” drop-down box (red arrow in Figure 11) on the toolbar above each BLAST hit to sort the alignment blocks based on different criteria (e.g., by E-value, query start position, subject start position). Each alignment block begins with a line that has the following format: “Range #:start to end” (where # is the alignment block number). You can use the “Next Match” and “Previous Match” links to navigate to the different alignment blocks within the same BLAST hit.
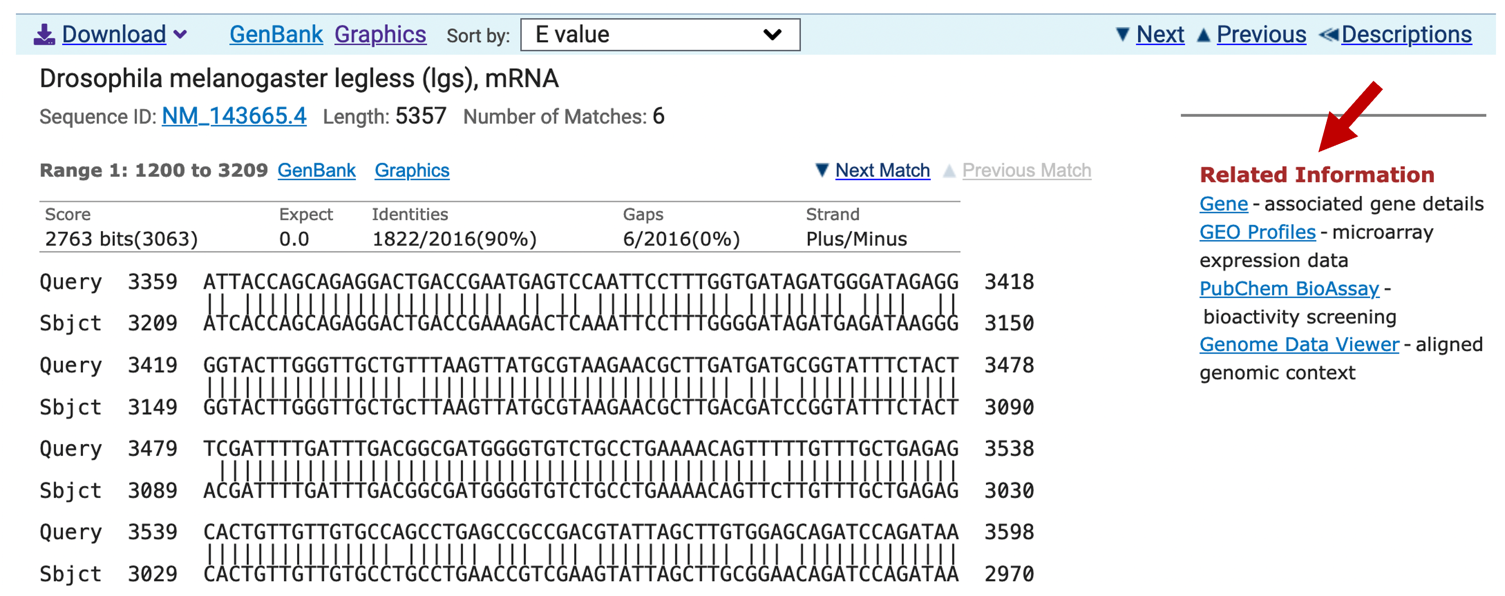
Depending on the database you use, there might be additional links to other parts of NCBI listed under the “Related Information” panel next to the sequence alignments. For example, there are links to Entrez Gene, GEO Profiles, and the Genome Data Viewer for the “Drosophila melanogaster legless (lgs), mRNA” (NM_143665.4; Figure 12). Entrez Gene provides us with an overview of the gene and links to literature references. GEO Profiles allow us to access expression data associated with the gene. The Genome Data Viewer allow us to view the BLAST alignments in a genome browser with other evidence tracks (e.g., gene annotations, RNA-Seq data, repeats).
What about the alignments themselves? Each alignment block begins with a summary, including the Expect value (i.e., E-value, or the statistical significance of the alignment), sequence identity (number of identical bases between the query and the subject sequence), the number of gaps in the alignment, and the orientation of the query relative to the subject sequence. The alignment consists of three lines: the query sequence, the matching sequence, and the subject sequence (Figure 13).
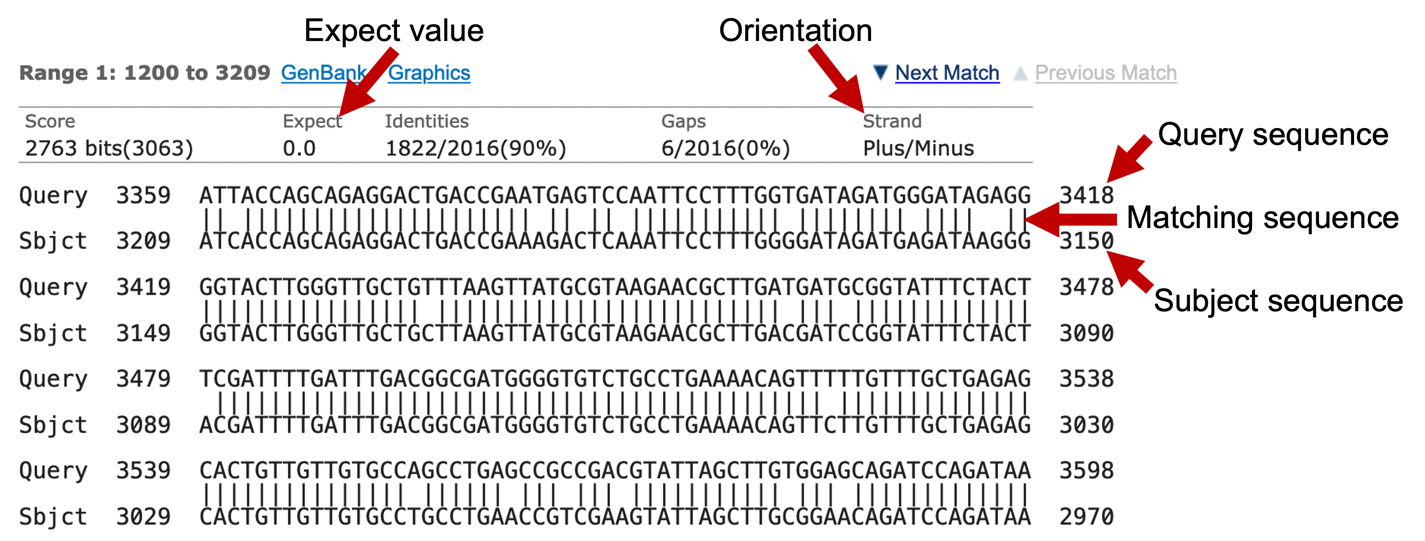
The - character in either the query or the subject sequence denotes a gap in the alignment (Figure 14).

By default, NCBI BLAST automatically masks low complexity sequences in the query sequence.
Depending on your BLAST search settings, these masked bases may appear as either grey lowercase letters (Figure 15) or as X’s.
The matching sequence consists of a combination of | and empty spaces, where | denotes a matching base between the query and subject sequences and the empty space denotes a mismatched base.

IV. Taxonomy
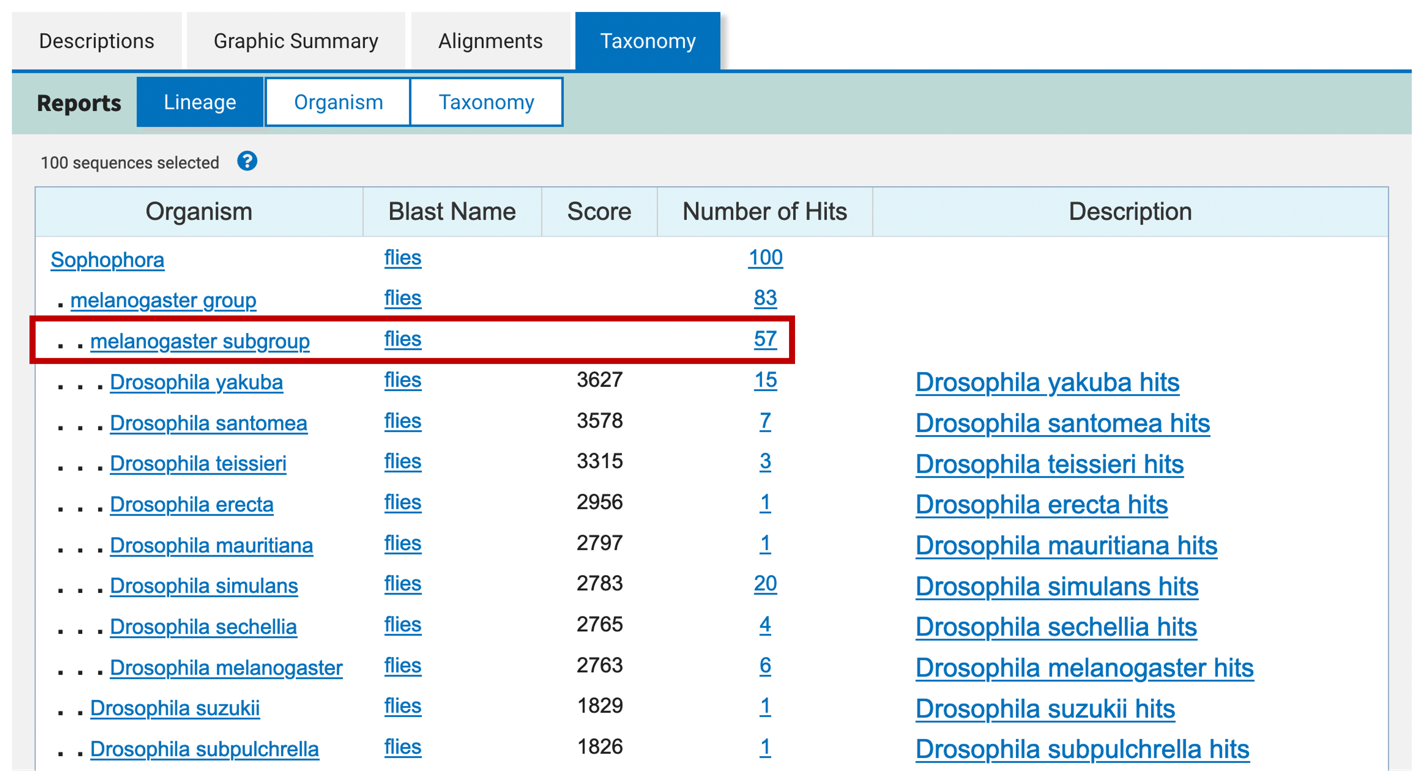
This tab shows the taxonomy of the selected BLAST hits in the Descriptions tab. The Taxonomy tab organizes the selected BLAST hits in three different report formats: Lineage, Organism, and Taxonomy (Figure 16).

The Lineage report provides an overview of the number of selected BLAST hits that are at each taxonomic level. The level of indentation in the “Organism” column corresponds to the taxonomic level. The value in the “Score” column corresponds to the maximum score for the BLAST hits of a terminal node. The value in the “Number of Hits” column shows the number of selected hits that are at the corresponding taxonomic level (Figure 17).
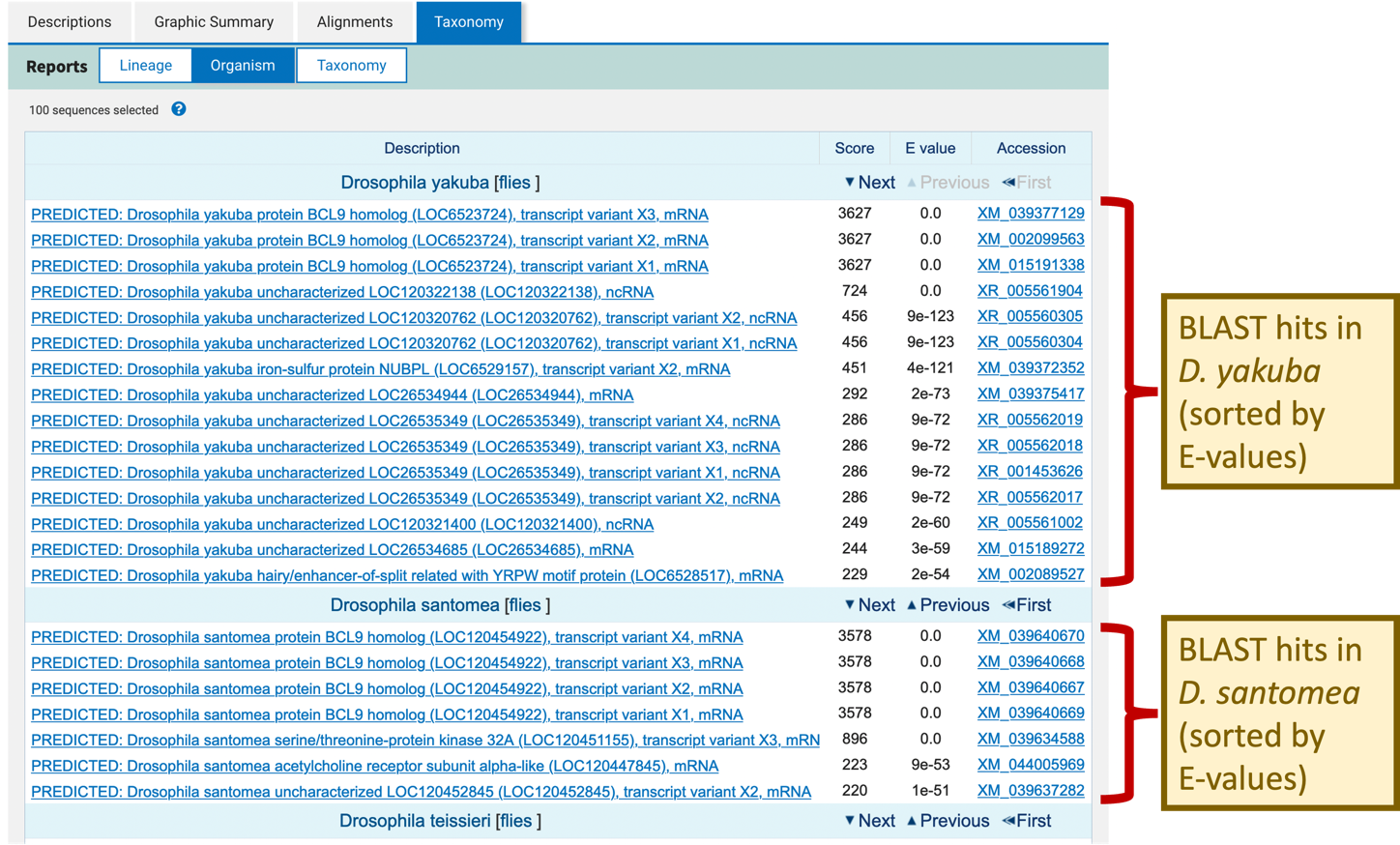
The Organism report groups the selected BLAST hits by organism. The BLAST hits for the different species are separated by a blue header. Within each species, the BLAST hits are sorted by E-value in ascending order (Figure 18).
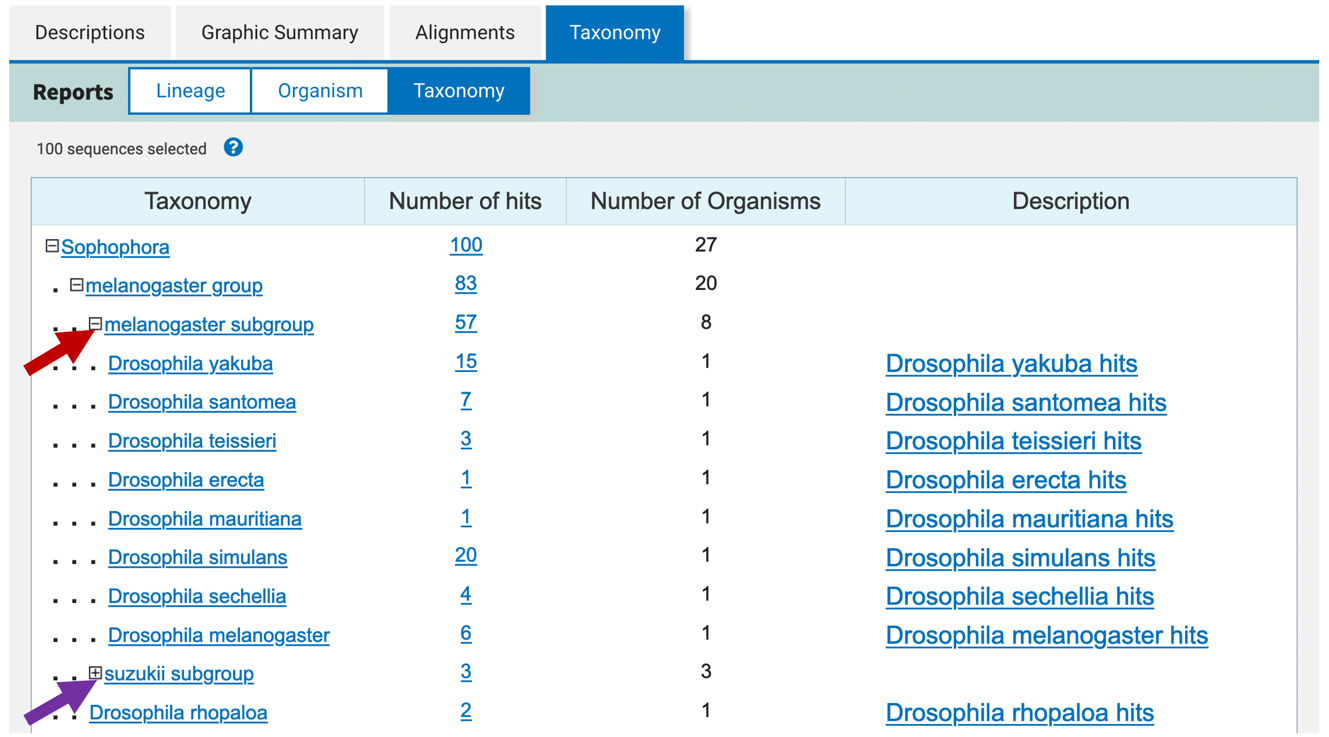
The Taxonomy report has a similar layout compared to the Lineage report. However, the Taxonomy report provides additional controls (the +/- icons under the “Taxonomy” column) to expand or collapse the non-leaf nodes (Figure 19). It also includes the number of organisms with BLAST hits at each taxonomic level.
Interpreting the blastn search result
Now that we have a better understanding of how the BLAST report is organized, we are ready to interpret the blastn results. The “Descriptions” and the “Graphic Summary” tabs (Figure 6 and Figure 8) show that many of the top hits are much more significant (with E-values of 0.0) than the rest of the blastn hits. Most of these top hits contain regions of sequence similarity that span the entire length of the query sequence (Figure 8). Looking at the descriptions and the corresponding GenBank records, it appears that these blastn hits correspond to the gene legless (also known as BCL9) in different Drosophila species.
Which of the hits under “Description” is the best hit for your unknown sequence (provide its name and accession number)? Use the metrics provided in the search results to support your reasoning.
|
|
We will ignore all computationally predicted genes and focus only on experimentally verified genes. This is because the experimentally verified genes are the only ones that we can be sure actually exist. |
From the blastn hit list, click on the description that corresponds to the best hit to an experimentally verified gene to navigate to the alignment between this hit and the unknown sequence.
What is the total length (number of base pairs) of the D. melanogaster legless mRNA?
Fill in Table 1 of the worksheet with information for each alignment block from the blastn search. Make sure to include the total number of mRNA base pairs in the table title.
|
|
If you do not have enough ranges to fill all rows, leave the remaining rows empty. Make sure your ranges are sorted by subject position. |
| Subject Position Range | Query Position Range | E-value | % Identity |
|---|---|---|---|
Use data from Table 1 to assert whether the entire D. melanogaster legless mRNA aligns to our unknown sequence. That is, provide evidence to support or reject whether the sequences matching D. melanogaster legless mRNA in your unknown sequence show collinearity.
Write a hypothesis indicating what gene from what species is homologous to a region of your D. yakuba unknown sequence.
Finding similar protein sequences using blastx
While it is useful to examine the mRNA sequence, often we are specifically interested in the protein products of the gene. Most RefSeq mRNA records include both translated and untranslated regions (i.e., the 5' and 3' UTRs). If we are interested in the protein, we would need to determine the location of the Coding DNA Sequences (CDS). Because every mRNA in the RefSeq RNA database has a corresponding sequence in the RefSeq Protein database, we will search our D. yakuba nucleotide sequence against the RefSeq Protein (refseq_protein) database with blastx. We now have all the information we need to setup the blastx search.
-
Navigate to the NCBI BLAST home page and click on the “blastx” image
-
Under the “Enter Query Sequence” section, click on the “Browse” or the “Choose File” button and select our sequence (unknown.fna).
-
Enter the Job Title “blastx search D. yakuba / RefSeq Protein”
-
In the “Choose Search Set” section, change the database to “Reference proteins (refseq_protein)”.
-
Check the box “Show results in a new window” next to the “BLAST” button
-
Click “BLAST” (Figure 20)
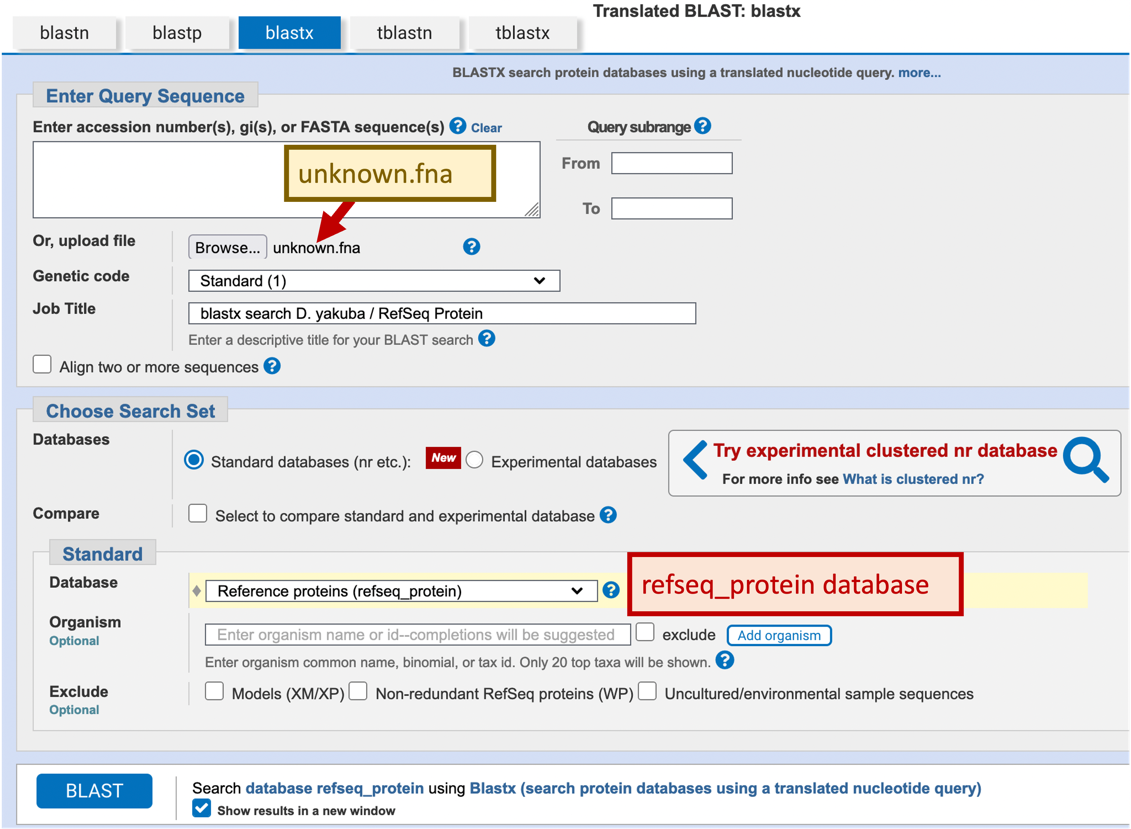
For teaching purposes, the blastx search result (blastxRefSeqProtein.txt) is available in the package for this exercise. (Note that this blastx search can take several minutes to complete.)
The blastx report is similar to the blastn report. It consists of the “Descriptions”, “Graphic Summary”, “Alignments”, and “Taxonomy” tabs. The “Graphic Summary” tab shows the highly significant hits to the legless protein in D. melanogaster and similar proteins in other Drosophila species. It also shows a few significant hits to transposases in the region between 6000-8000 bp of our sequence (Figure 21).
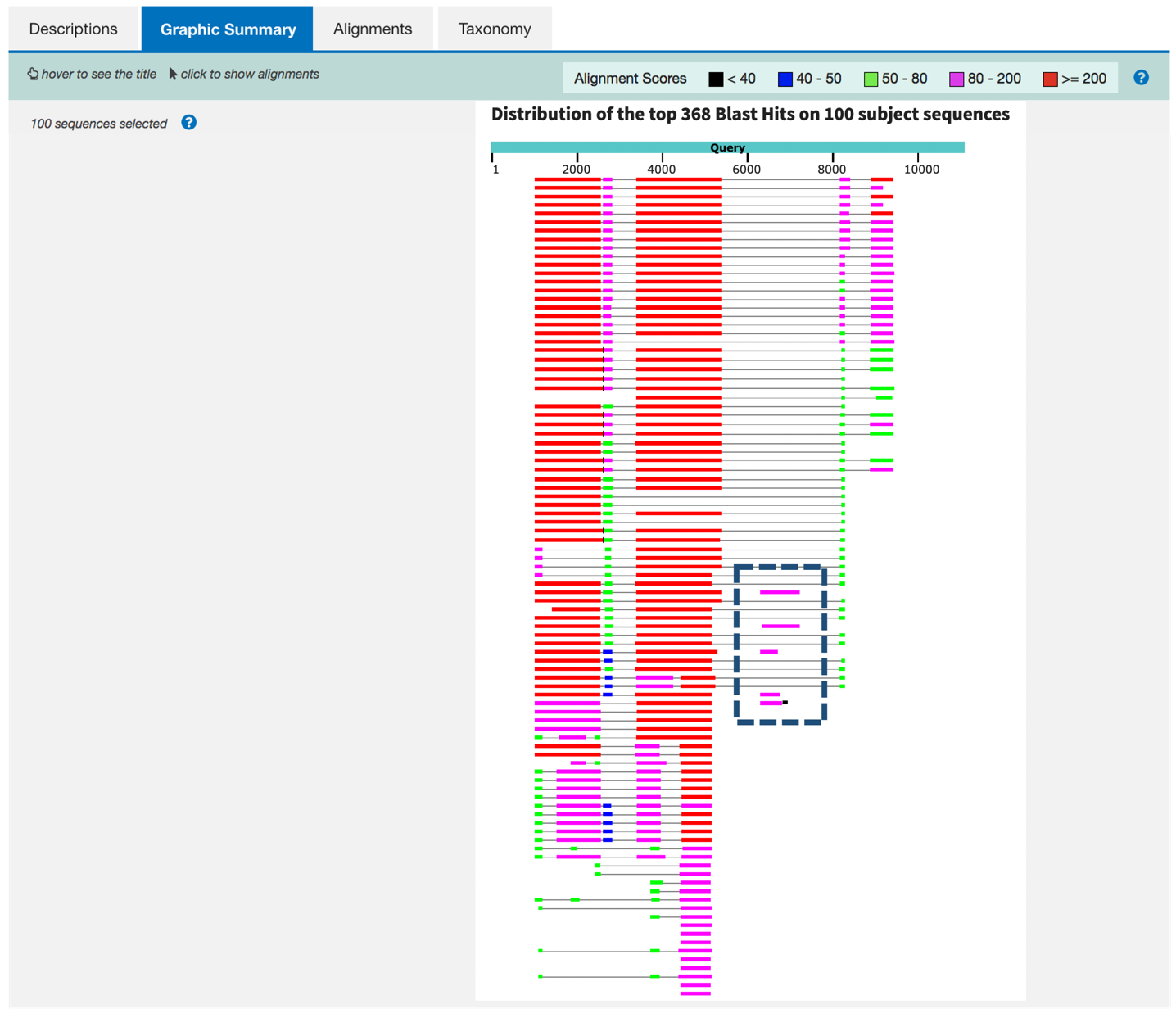
These hits suggest our sequence contains a type of repetitious element called a transposable element. In future exercises, we will learn how we can reduce the number of spurious hits in our BLAST reports by masking these elements prior to performing the BLAST search. For now, we will ignore these additional matches and focus on the best manually curated RefSeq hit — the D. melanogaster legless protein (NP_651922.1; Figure 22).
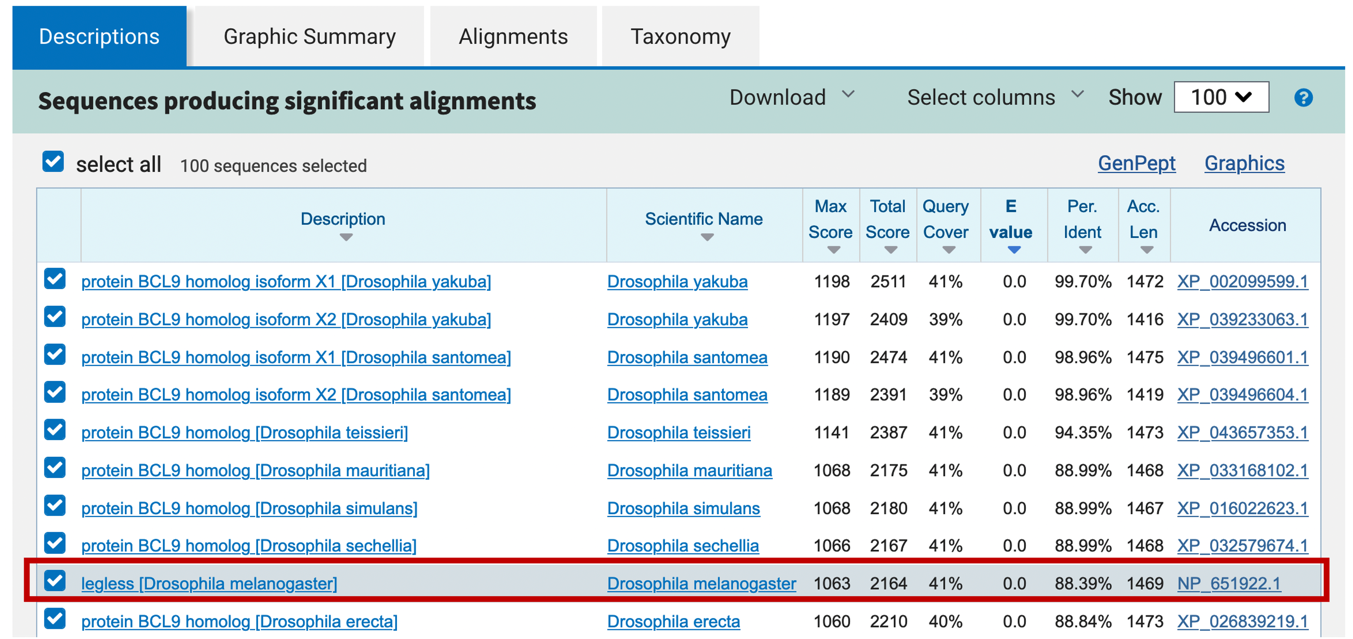
How do RefSeq records with the “XM_” prefix differ from records with the “NM_” prefix? How do RefSeq records with the “NM_” prefix differ from records with the “NP_” prefix?
We can analyze the blastx alignments in the same way that we have previously analyzed the blastn report. However, because blastx translates the input sequence in all 6 reading frames before comparing our sequence with the protein database, there is an additional “Frame” field in each alignment block. The frame begins with either + or -, which corresponds to the relative orientation of our sequence compared to the protein. The number following the relative orientation in the frame field ranges from 1 to 3, which reflects the reading frame that produces the translated peptide sequence. Collectively, the relative orientation and the number can be used to represent all 6 reading frames. A frame shift between two alignment blocks in the blastx match often indicates that the two alignment blocks correspond to different coding exons. The “Positives” field corresponds to the number of amino acids that are either identical or have similar chemical properties between the translated query and the subject sequences (Figure 23).
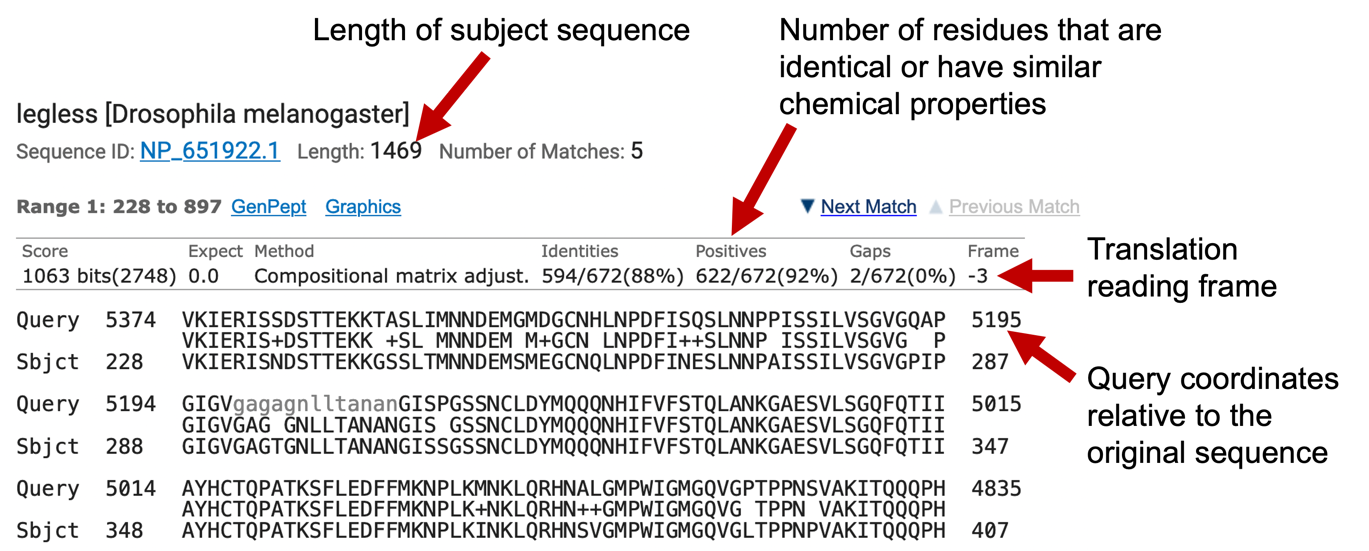
Similar to the blastn alignment, each alignment block in our blastx report also consists of three lines: the query sequence, the matching sequence, and the subject sequence. Note that the query sequence has been translated into the corresponding amino acid sequence in the reading frame specified by the “Frame” field. However, the coordinates of the query sequence are still relative to the original nucleotide sequence. Like our blastn alignment, the grey lowercase residues in the query sequence correspond to low complexity sequences that were masked by BLAST.
There are some minor differences in the matching sequence of the blastn and blastx outputs. Residues in the matching sequence represent amino acids that are identical between the query and subject sequences. The “+” character denotes amino acids that are different between the query and subject, but these different amino acids have similar chemical properties. A space indicates that the two aligned amino acid in the query and subject are different, and they have different chemical properties.
What is the total length (number of amino acids) of the D. melanogaster legless protein?
Fill in Table 2 of the worksheet with information for each alignment block from the blastx search.
|
|
If you do not have enough ranges to fill all rows, leave the remaining rows empty. Make sure your ranges are sorted by subject position. |
| Subject Position Range | Query Position Range | E-value | % Identity | % Positives | Frame |
|---|---|---|---|---|---|
Use data from Table 2 of your worksheet to assert whether the entire D. melanogaster legless protein aligns to your unknown sequence. First add what support you find, using similar reasoning as in Question 3. Then note any discrepancies, clearly distinguishing between supporting and conflicting evidence.
Using bl2seq to compare coding sequences of a gene to a sequence
The blastn and blastx search results suggest that we have identified the putative ortholog of the legless gene in our D. yakuba sequence. However, in order to construct a complete gene model, we must resolve the discrepancies in the alignments of our blastn and blastx output. Because coding sequences are under strong selective pressure and are more likely to be conserved than other regions of the genome, our first step is to identify the coding sequences of our putative gene. To begin the more detailed analysis, we will perform a series of BLAST searches using the amino acid sequence of each exon in the D. melanogaster version of the legless gene. It will be helpful to our annotation efforts if we can obtain the amino acid sequence that corresponds to each exon individually. Fortunately, we can easily obtain the individual exon sequences using the Gene Record Finder.
-
Open a new web browser tab and navigate to the Gene Record Finder website.
-
Type “lgs” (the official FlyBase symbol for the legless gene) in the textbox and click on the “Find Record” button (Figure 24)
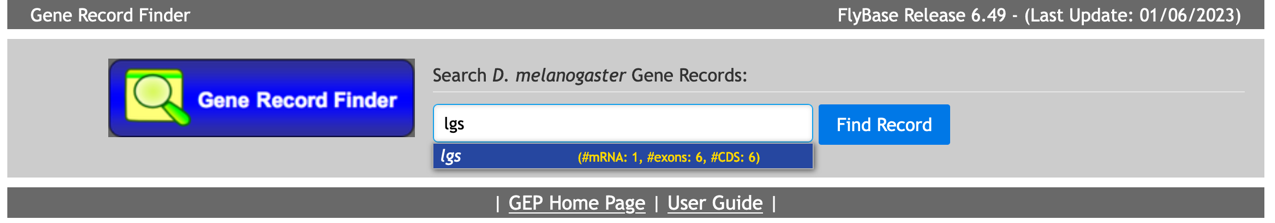
In the “mRNA Details” section of the gene report, we notice that there is only one isoform of the legless gene in D. melanogaster (lgs-RA, the A isoform of lgs). We can access all the transcribed exons through the “Transcript Details” tab and all the coding sequences (CDS) through the “Polypeptide Details” tab (Figure 25).
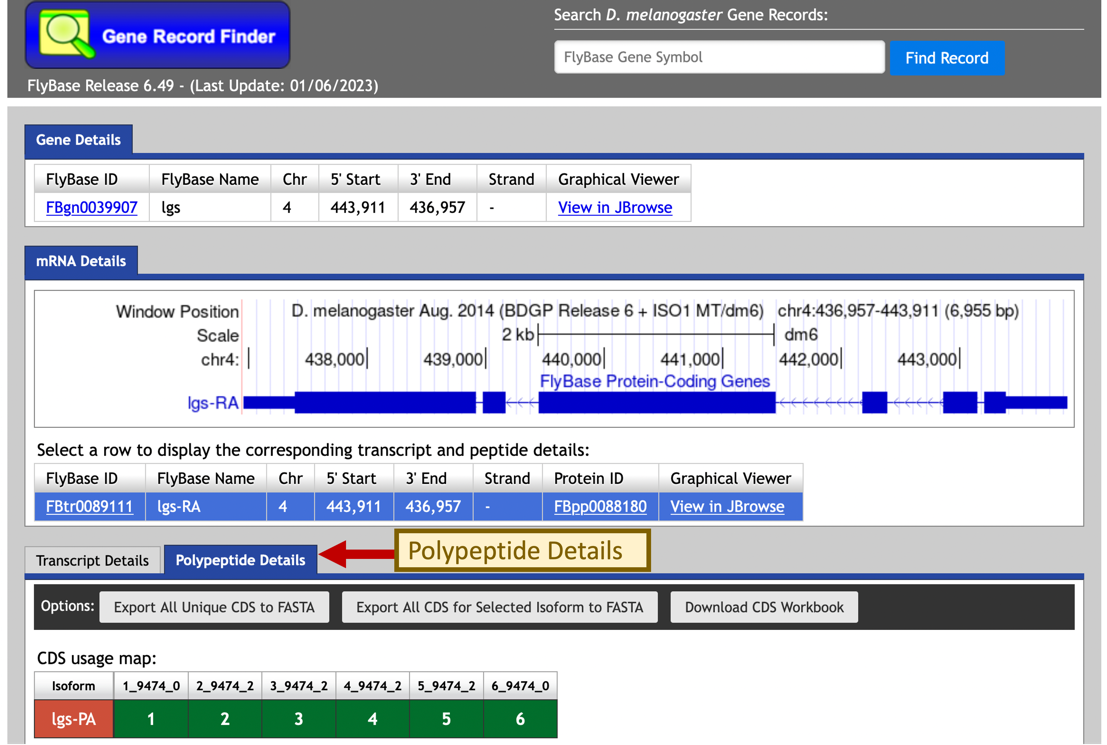
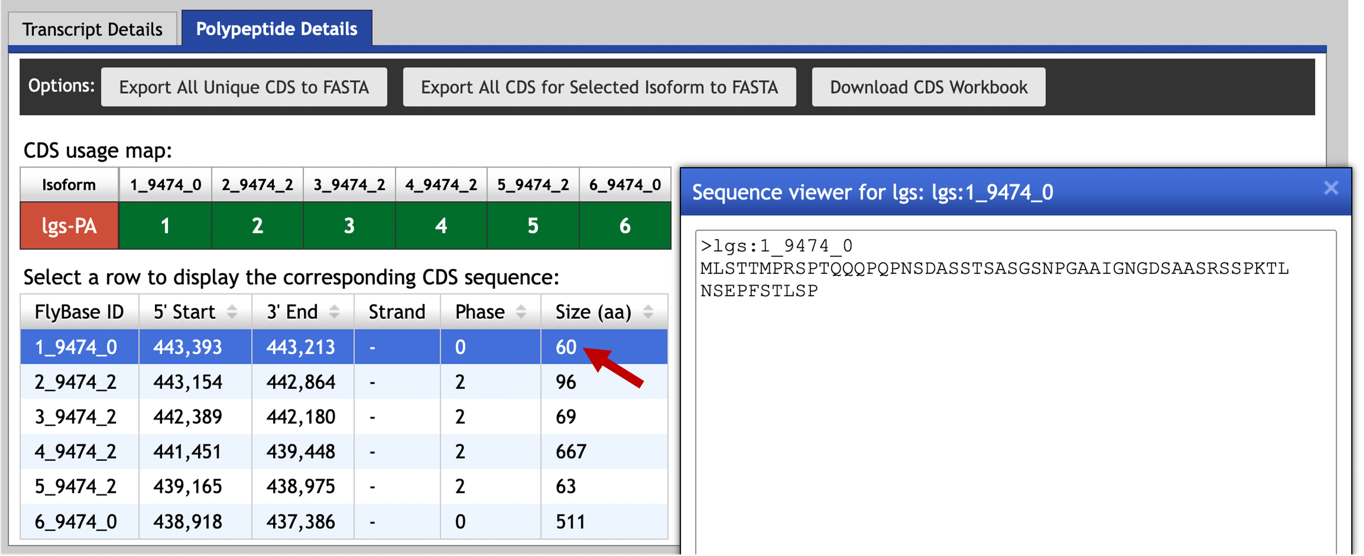
To retrieve the amino acid sequence for each coding sequence, click on the row that corresponds to the coding sequence in the CDS table (Figure 26).
To determine the locations of the coding sequences, we will perform BLAST searches to compare the individual exons from D. melanogaster with our D. yakuba sequence. Because we are comparing a protein sequence against a nucleotide sequence, we will use the tblastn program for our search. In order to prevent BLAST from masking low complexity regions in our protein, we will turn off the low complexity filter. In addition, because we are only comparing two sequences, we will also turn off compositional adjustments under scoring parameters.
-
Select the first CDS (1_9452_0) from the Gene Record Finder CDS table and copy the sequence to the clipboard
-
Open a new web browser tab and navigate to the NCBI BLAST home page; click on the “tblastn” image under the “Web BLAST” section
-
Select the checkbox “Align two or more sequences” under the “Enter Query Sequence” section
-
Paste the CDS sequence for 1_9452_0 into the “Enter Query Sequence” field
-
For the “Subject Sequence”, click on the “Browse” or the “Choose File” button and select our unknown sequence (unknown.fna)
-
Click on the “Algorithm parameters” link to expand this section. Verify that the “Word size” parameter is set to 3.
-
Change the “Compositional adjustments” field to “No adjustment” under the “Scoring Parameters” section
-
Uncheck the box “Low complexity regions” under “Filters and Masking”
-
Click “BLAST” (Figure 27).
For teaching purposes, the BLAST output (bl2lgsExon1_tblastn.txt) is available in the package for this exercise.
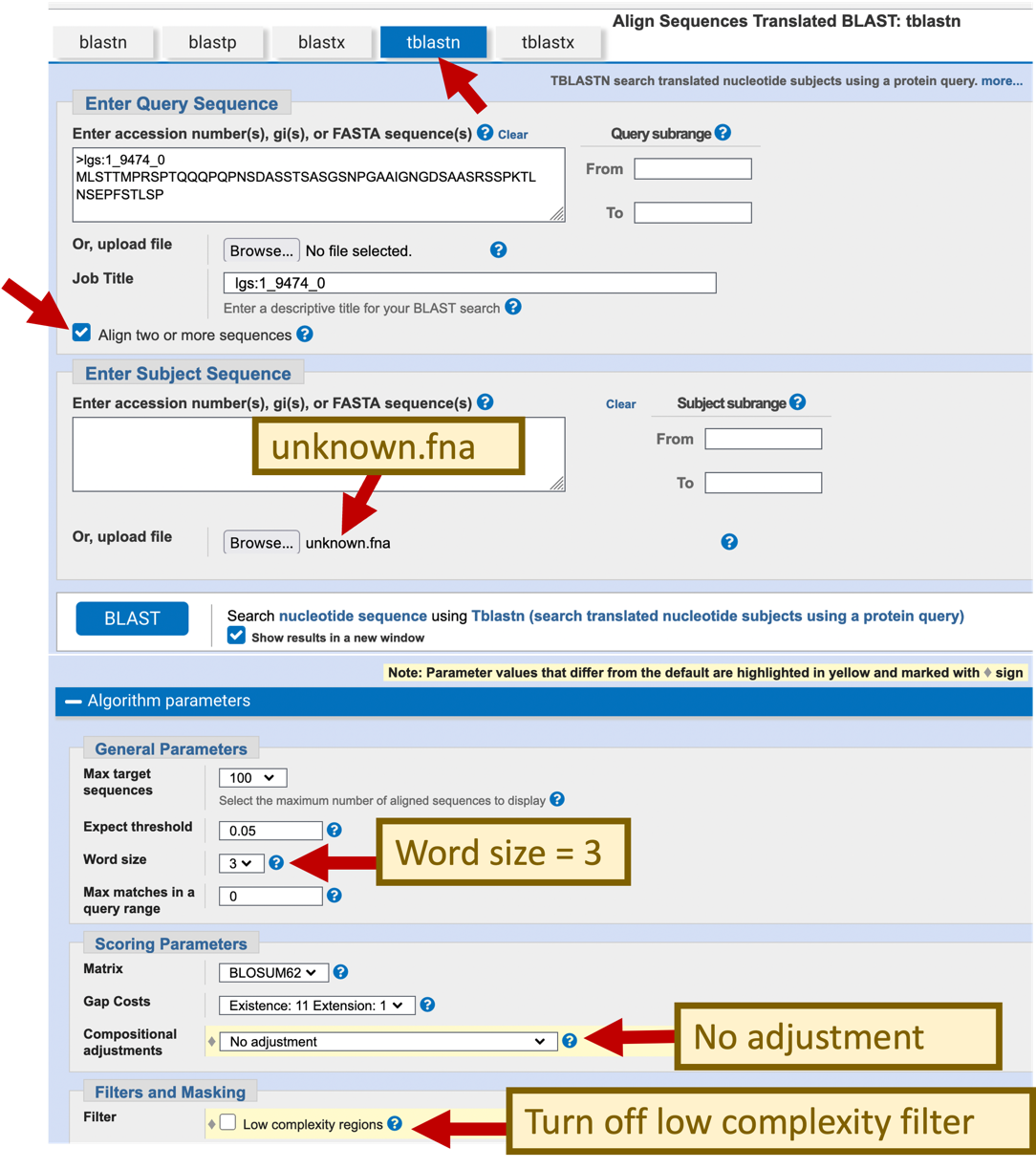
The “Alignments” tab of the tblastn output shows that the first coding exon has a length of 60 amino acids, and it aligns to 9344-9168 of the query sequence when it is translated in frame -2 (Figure 28).
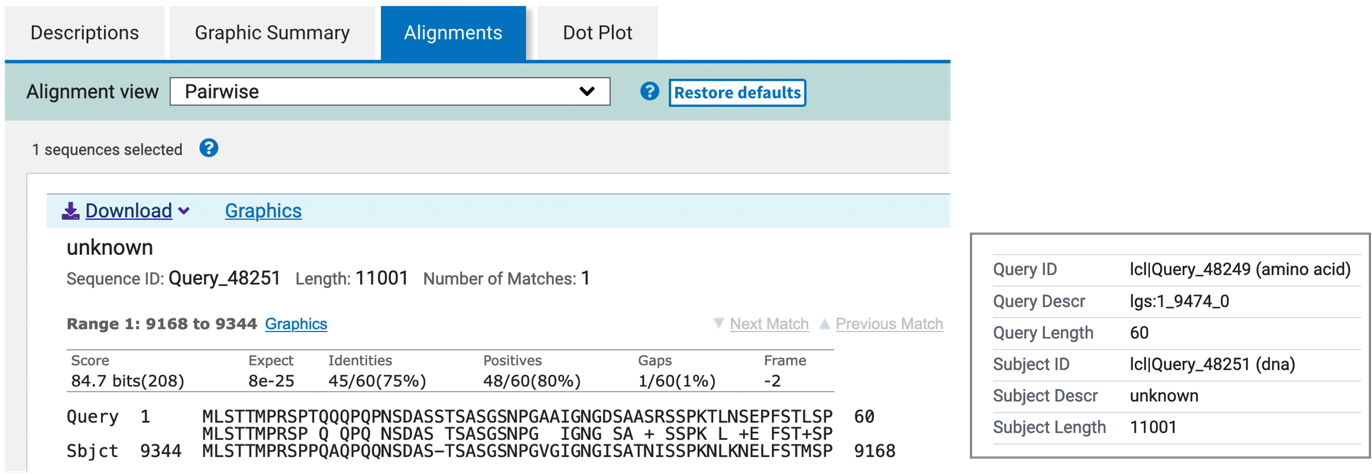
We can use the same strategy to map the rest of the coding exons.
Perform tblastn searches of each CDS of legless (query) against the unknown sequence (subject). Fill in Table 3 on your worksheet based on the tblastn search results.
| CDS # | FlyBase ID | Query Position Range | Subject Position Range | E-value | % Identity | % Positives | Frame |
|---|---|---|---|---|---|---|---|
Use data from Table 3 to explain whether all CDS of the D. melanogaster legless gene are accounted for in your unknown sequence. Explain how the CDS tblastn alignments resolve the discrepancies discussed in Question 7.
Additional exercise: Define the 5' UTR of legless using blastn
We could apply the same strategy to map the locations of the putative untranslated regions (UTRs) using bl2seq with the blastn program. Go back to the Gene Record Finder web browser window. The genome browser image in the “mRNA Details” panel shows that the first CDS of legless (1_9452_0) is part of a larger exon (lgs:1) that includes the 5' UTR (Figure 29). To estimate the location of the 5' UTR in our unknown sequence, we will perform a blastn search of the exon lgs:1 against the unknown sequence, and then compare the alignment with the tblastn search result for CDS 1_9452_0.
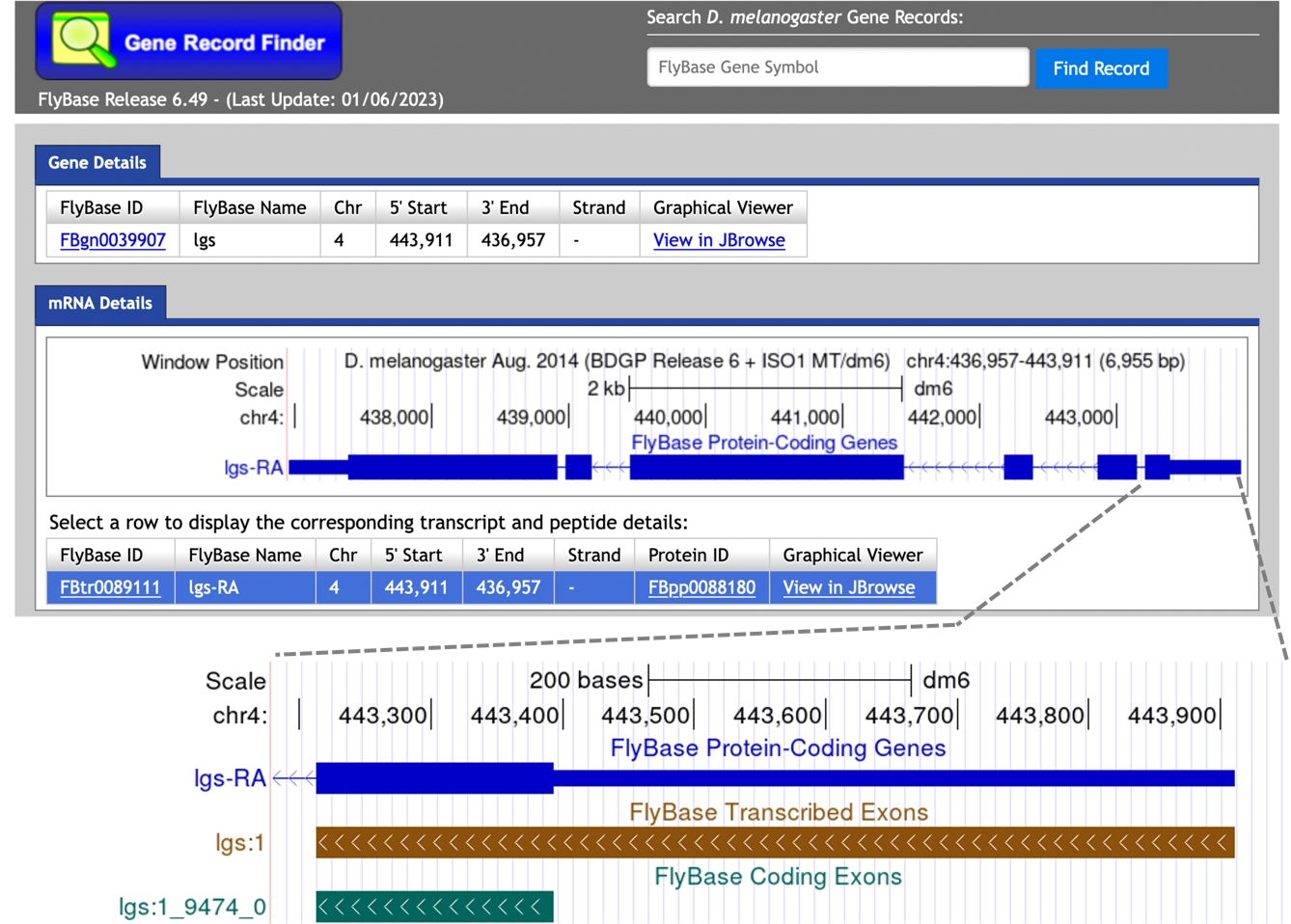
To retrieve the exon sequence for the first transcribed exon, select the “Transcript Details” tab in the Gene Record Finder and then click on the first row in the exon table (Figure 30).
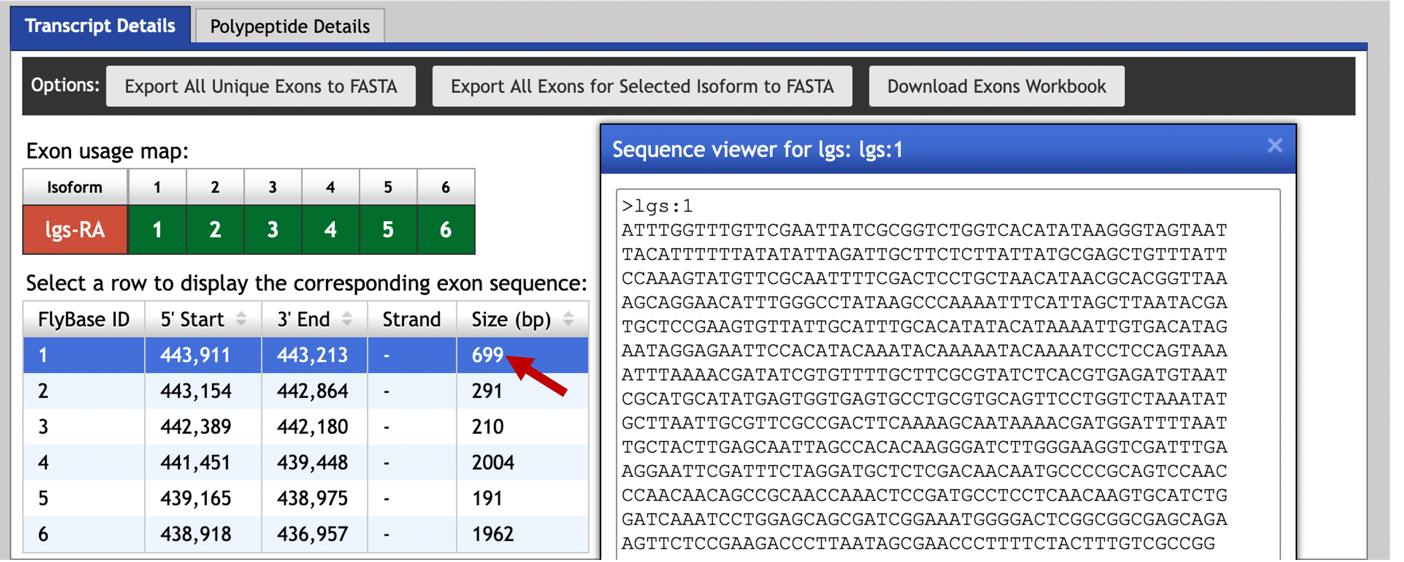
We can then use the following steps to perform the blastn search:
-
Select the first exon (lgs:1) from the Gene Record Finder exon table and copy the sequence to the clipboard
-
Open a new web browser tab and navigate to the NCBI BLAST home page; click on the “Nucleotide BLAST” image under the “Web BLAST” section
-
Select the checkbox “Align two or more sequences” under the “Enter Query Sequence” section
-
Paste the exon sequence for lgs:1 into the “Enter Query Sequence” field
-
For the “Subject Sequence”, click on the “Browse” or the “Choose File” button and select our unknown sequence (unknown.fna)
-
Select the “Somewhat similar sequences (blastn)” option under “Program Selection”
-
Click on the “Algorithm parameters” link to expand this section.
-
Uncheck the box “Low complexity regions” under “Filters and Masking”
-
Click “BLAST” (Figure 31).
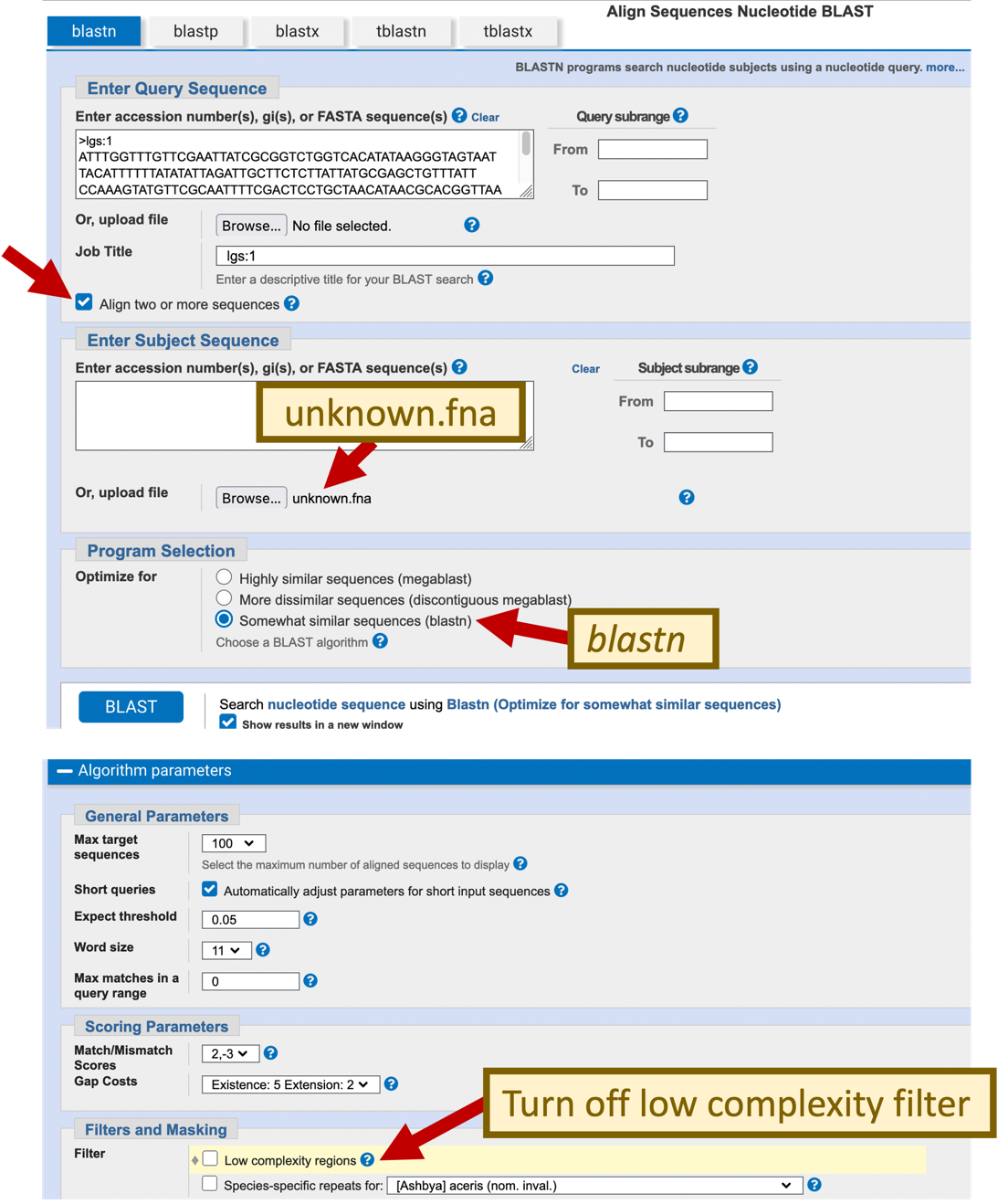
For teaching purposes, the BLAST output (bl2lgsExon1_blastn.txt) is available in the package for this exercise.
Based on the blastn search result, where is the best match for lgs:1 in the unknown sequence? What are the E-value and the percent identity for this blastn alignment? Based on the tblastn result for CDS 1_9452_0 and the blastn alignment for lgs:1, where is the 5' UTR for lgs in the unknown sequence?
Conclusion
In this exercise, we have used multiple BLAST programs to identify and characterize a putative gene in a genomic sequence from D. yakuba. You are now ready to tackle some of the more challenging BLAST exercises on the GEP website: