Prerequisites
Resources
Files for this walkthrough
The package containing the files for this walkthrough are available through the "Motif Discovery in Drosophila" page on the GEP website.
Introduction
Interactions between proteins and DNA are one of the main mechanisms for regulating chromosome function and gene expression. A subset of DNA binding proteins, including the transcription factors, exhibits sequence-specific affinity. Consequently, short conserved motifs are often found near transcription start sites (TSS), corresponding to sites where transcription factors bind to the DNA. These interactions generally help regulate the expression of nearby genes, often in a tissue or developmental time point specific manner.
In this walkthrough, we will try to identify conserved motifs upstream of a group of the dot chromosome (also known as the Muller F element) genes in Drosophila melanogaster using publicly available Drosophila databases and genome analysis tools. Identifying such motifs will allow us to look for unique features in the regulation of dot chromosome genes. These genes function in an unfavorable (heterochromatic) environment, and so may require additional signals (multiple copies, an enhanced cluster, etc.) for activation.
Identify genes with similar expression patterns
The first step in our analysis is to identify a set of genes in D. melanogaster with similar gene expression patterns; these genes are more likely to be under the control of a similar set of transcription factors. The modENCODE project has previously generated a large set of high-throughput expression (RNA-Seq) data at different developmental stages, tissues, and cell lines. FlyBase has curated these RNA-Seq datasets and we can use the RNA-Seq search tools at FlyBase to identify genes with similar expression profiles.
Use FlyBase RNA-Seq tools to identify genes with similar expression patterns
Open a web browser and navigate to the FlyBase website. Click on the "RNA-Seq" image on the FlyBase home page and then click on the "RNA-Seq Profile" link (Figure 1) to access the "RNA-Seq Expression Profile Search" tool.
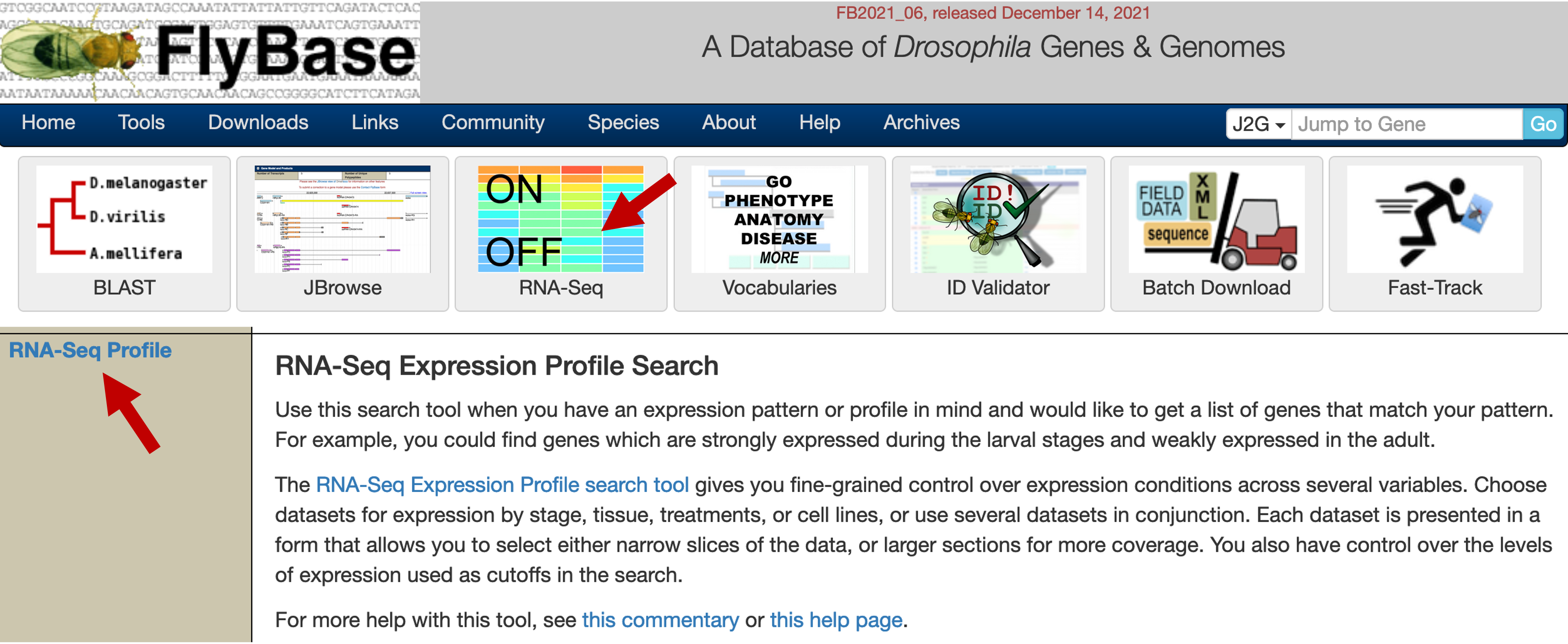
We can use this tool to identify genes that are expressed at specific developmental times, tissues, treatments, cell types, or any combinations thereof. By default, genes with low or no expression according to the modENCODE gene expression scale are considered to be "off" while genes with moderately high expression or above are considered to be "on" (see [Graveley et al., 2011] for details).
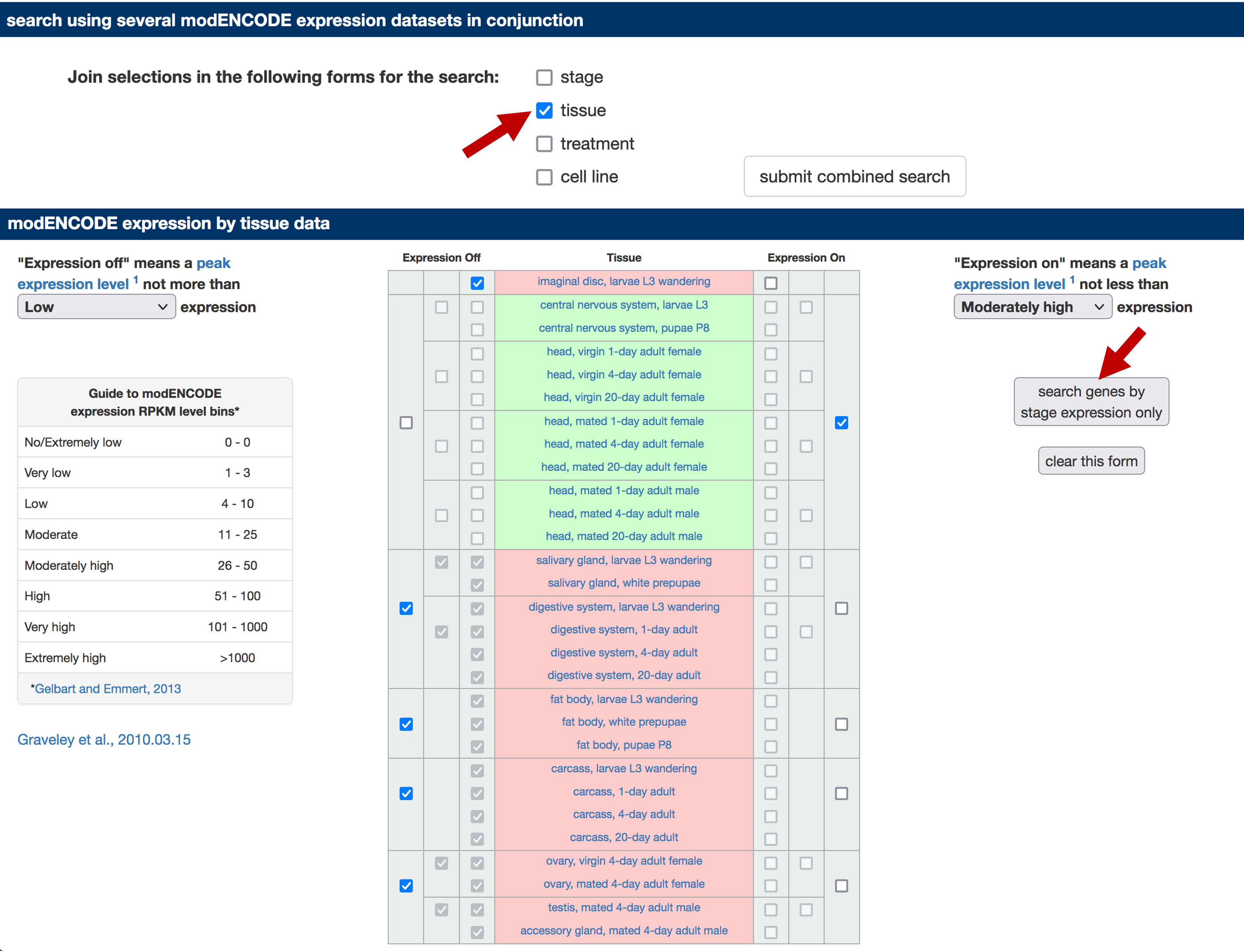
In this walkthrough, we will identify the subset of D. melanogaster genes that are expressed only in head tissues. Uncheck the "stage" checkbox under the "search using several modENCODE expression datasets in conjunction" section. Select the "Expression On" checkbox for all the head tissues and the central nervous system entries. Select the "Expression Off" checkbox for all the other tissues (Figure 2). Click on the "search genes by stage [sic] expression only" button to run the search.
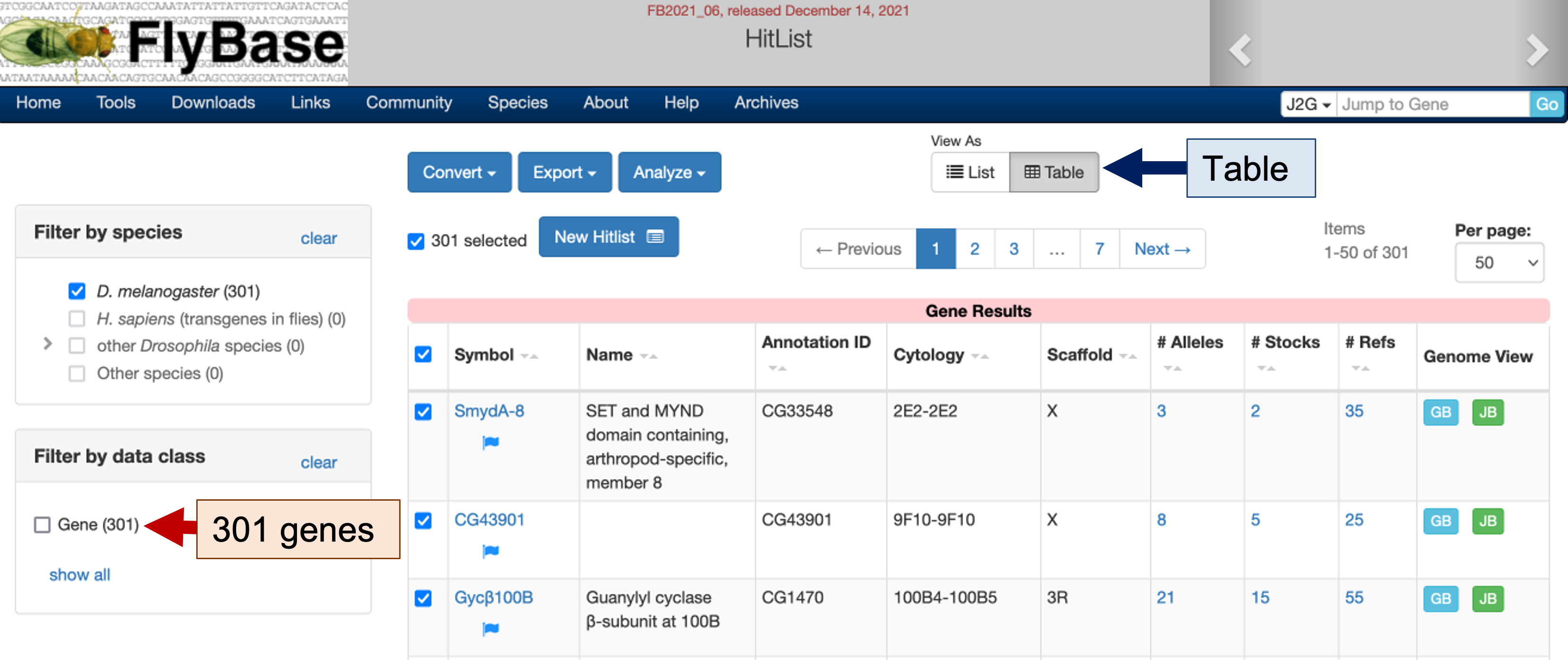
The search results page shows 301 D. melanogaster genes that are expressed only in head tissues (Figure 3). To view this list of genes in a more compact format, click on the "Table" button under the "View As" section (blue arrow in Figure 3). We can click on the links under the "Symbol" column to learn more about each gene (the blue flag next to the gene symbol indicates the record has changed recently). We can use the "Export" button to export the gene list and use the "Analyze" button to filter this list of genes.
Filtering the gene list by chromosomes
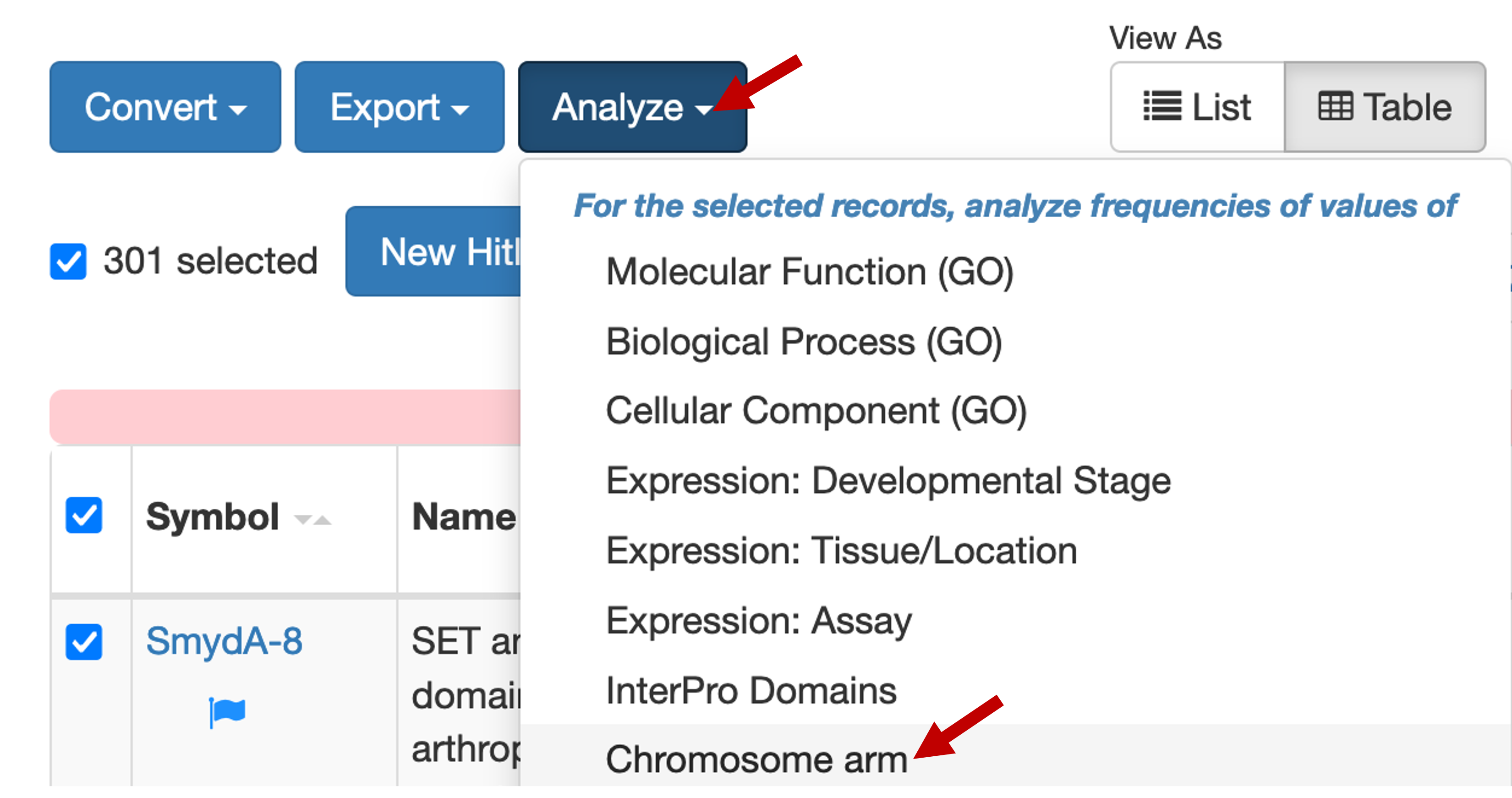
In this example, we would like to investigate the set of dot chromosome genes that are only expressed in head tissues. To filter this initial list of genes by chromosome, click on the "Analyze" button and select "Chromosome arm" in the drop-down menu (Figure 4).
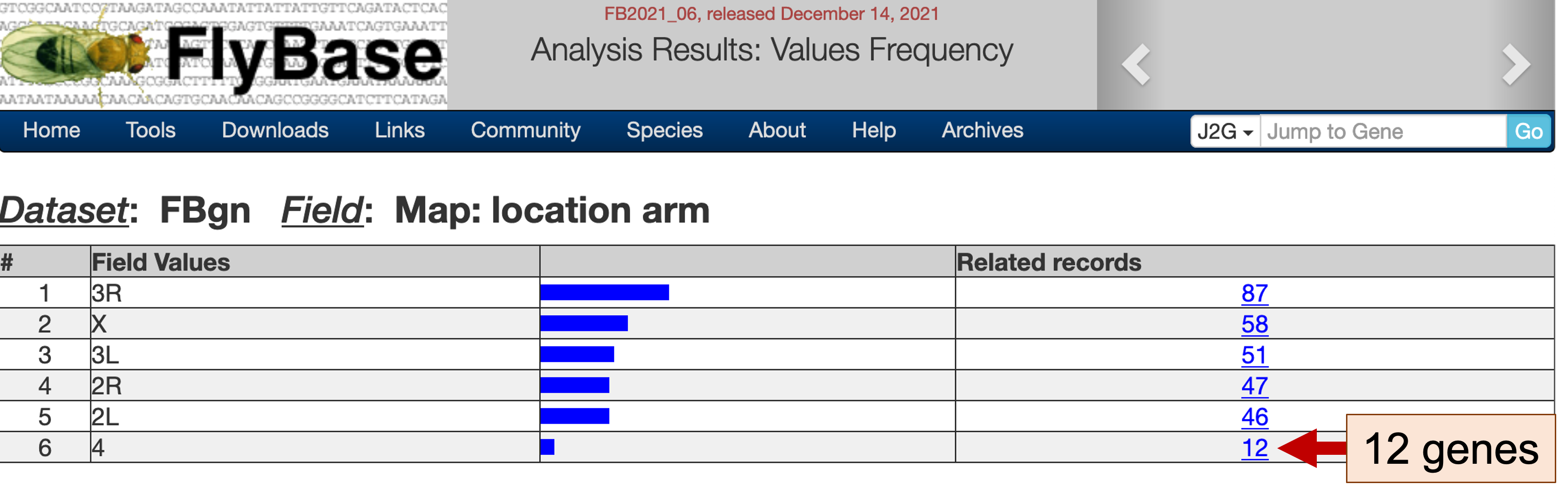
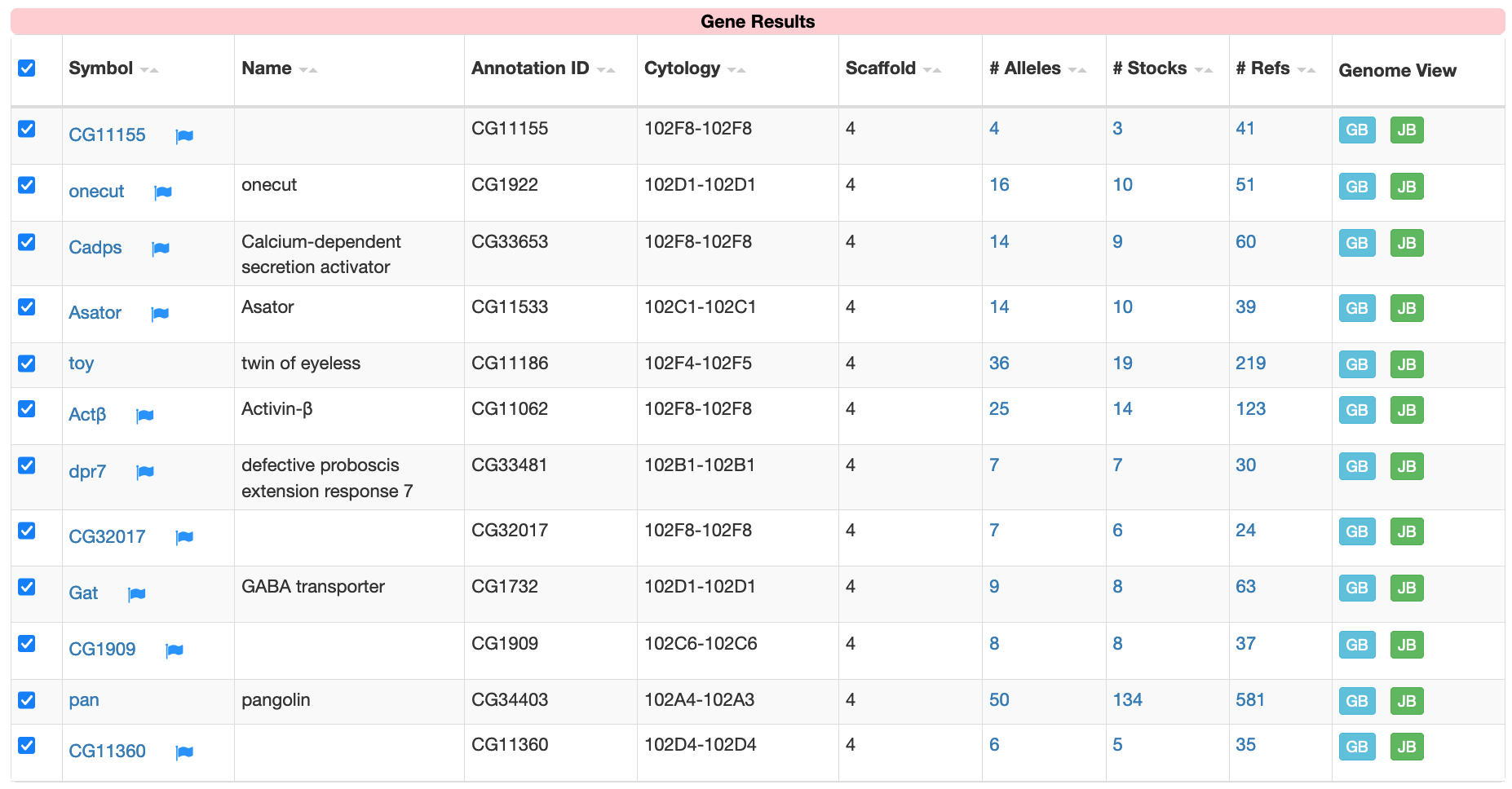
The "Values Frequency" page shows the distribution of the genes in the gene list among the different chromosomes. Under the "Related records" column, we see that 12 of the 301 genes that are expressed only in head tissues are found on the dot chromosome (Figure 5). Click on this link (with the label 12) to filter the original list of genes so that only dot chromosome genes are in the new gene list (Figure 6). We could also apply other filters to the gene list (e.g., using controlled vocabularies to filter the list by molecular functions or biological processes) to manipulate the list of gene candidates that we would like to use to discover common motifs.
Use sequence comparisons to identify conserved motifs
In this walkthrough, we will analyze this list of 12 dot chromosome genes that are expressed only in head tissues to ascertain if there are motifs upstream of the transcription start site (TSS) that appear at high frequencies.
Identify the regions upstream of the transcription start sites
The first step of this analysis is to retrieve the genomic sequences upstream of the TSS for each gene in the gene list. Specifically, we will extract the region from the start of the TSS of the gene of interest to the beginning of the adjacent gene. (Depending on your research goal, you can adjust the size of the search region. For example, many regulatory elements are found within 2kb upstream of the TSS.) The procedure used to retrieve the upstream sequences is described below. For teaching purposes, the file with the upstream sequences (dot_genes_head_expressed.fasta) is available in the package for this walkthrough.
Open a new tab and navigate to FlyBase. Enter the name of a gene in the gene list (e.g., Actbeta, which appears as "Actβ") into "Jump to Gene" (J2G) search box on the top right corner of the main navigation bar and then click "Go" (Figure 7).
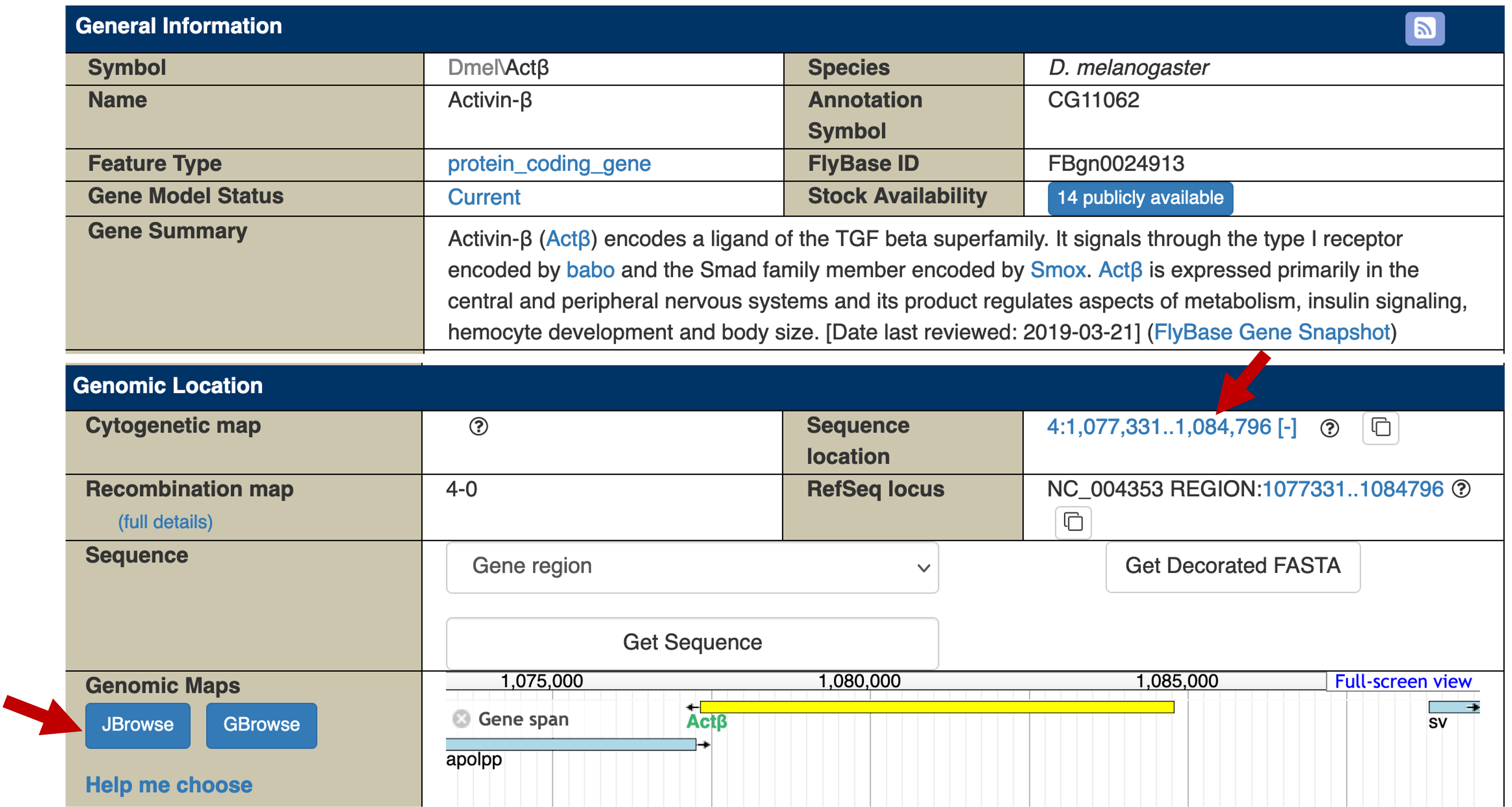
The JBrowse image under the "Genomic Location" section shows that the Actbeta gene is on the minus strand and the "Sequence location" field shows that this gene spans from 1,077,331-1,084,796 (Figure 8). Because this gene is on the minus strand, the first nucleotide before the TSS of Actbeta is located at 1,084,797.
The JBrowse image in the "Genomic Maps" section shows that the first gene upstream of the TSS of Actbeta is sv. To determine the precise coordinates of the sv gene, click on the "JBrowse" button (under "Genomic Maps") and then click on the large "Zoom out" icon in the toolbar (Figure 9).
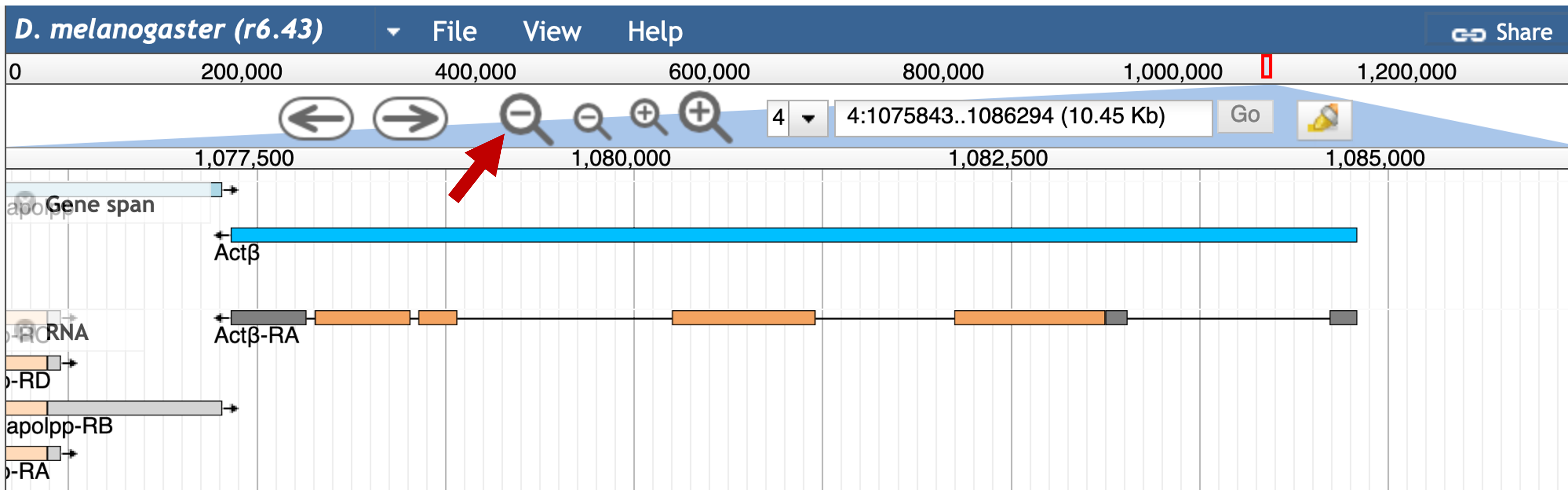
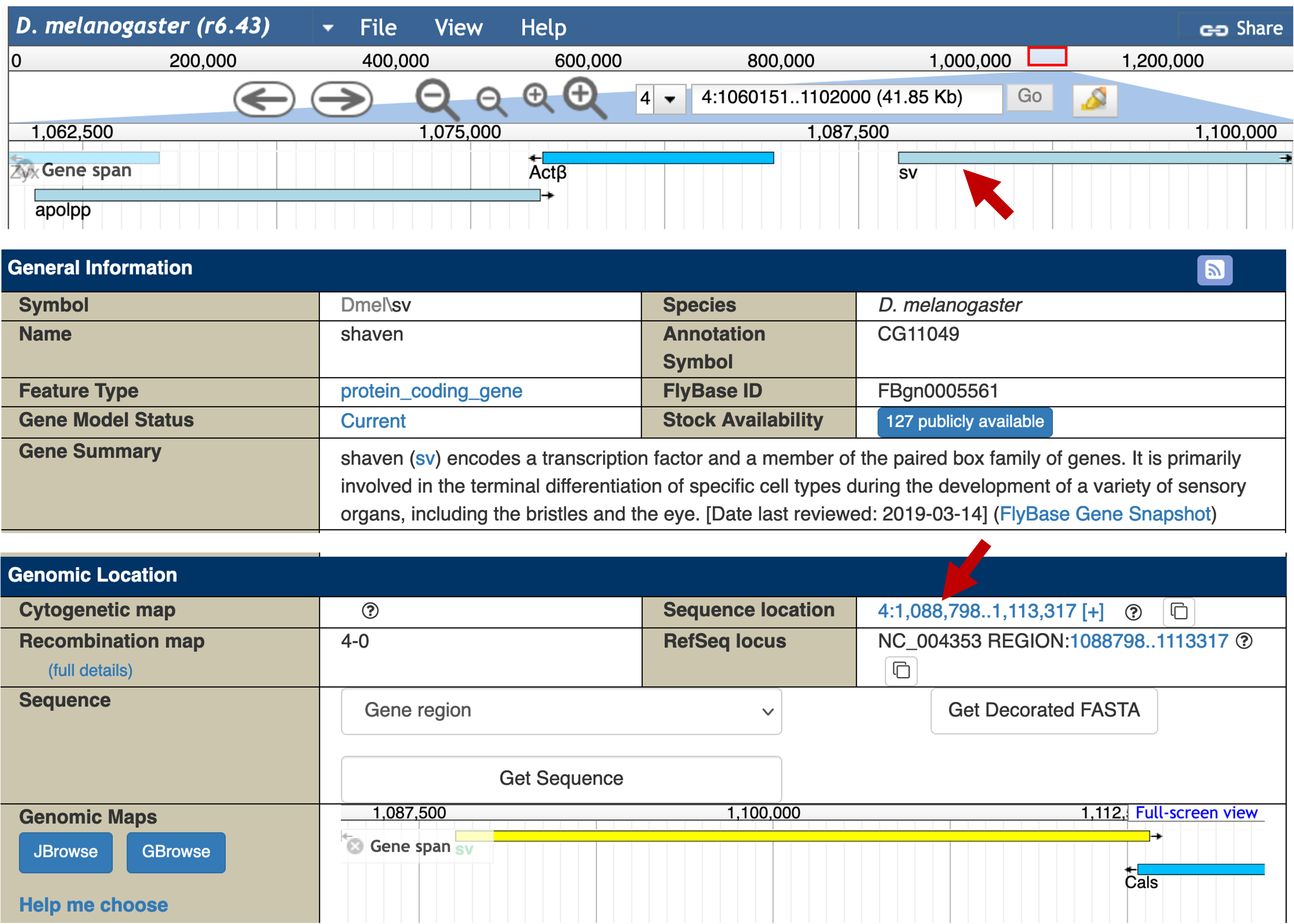
Click on the sv feature in the "Gene span" track. The FlyBase Gene Report shows that sv is on the plus strand and it begins at 1,088,798 (Figure 10). Consequently, the genomic coordinates for the region between the TSS of Actbeta and the beginning of sv is 4:1084797-1088797.
We can apply this protocol to the other genes in our gene list to determine the coordinates for all the upstream regions. The coordinates of all the upstream regions are listed below:
| Gene | Upstream region |
|---|---|
Actbeta |
4:1084797-1088797 |
Asator |
4:488995-501809 |
Cadps |
4:1230714-1230836 |
CG1909 |
4:580123-583107 |
CG11155 |
4:1125326-1126601 |
CG11360 |
4:669885-671141 |
CG32017 |
4:1162159-1164881 |
dpr7 |
4:253330-254996 |
Gat |
4:626032-629690 |
onecut |
4:607650-610683 |
pan |
4:67208-69325 |
toy |
4:978490-989724 |
Retrieve the upstream sequences
Once we have generated the list of coordinates for the upstream regions, we can use the FlyBase Sequence Downloader tool to retrieve the genomic sequences for these upstream regions. You can access this tool through the FlyBase main navigation bar (under "Tools" → "Query by symbols/IDs" → "Sequence Downloader", Figure 11).
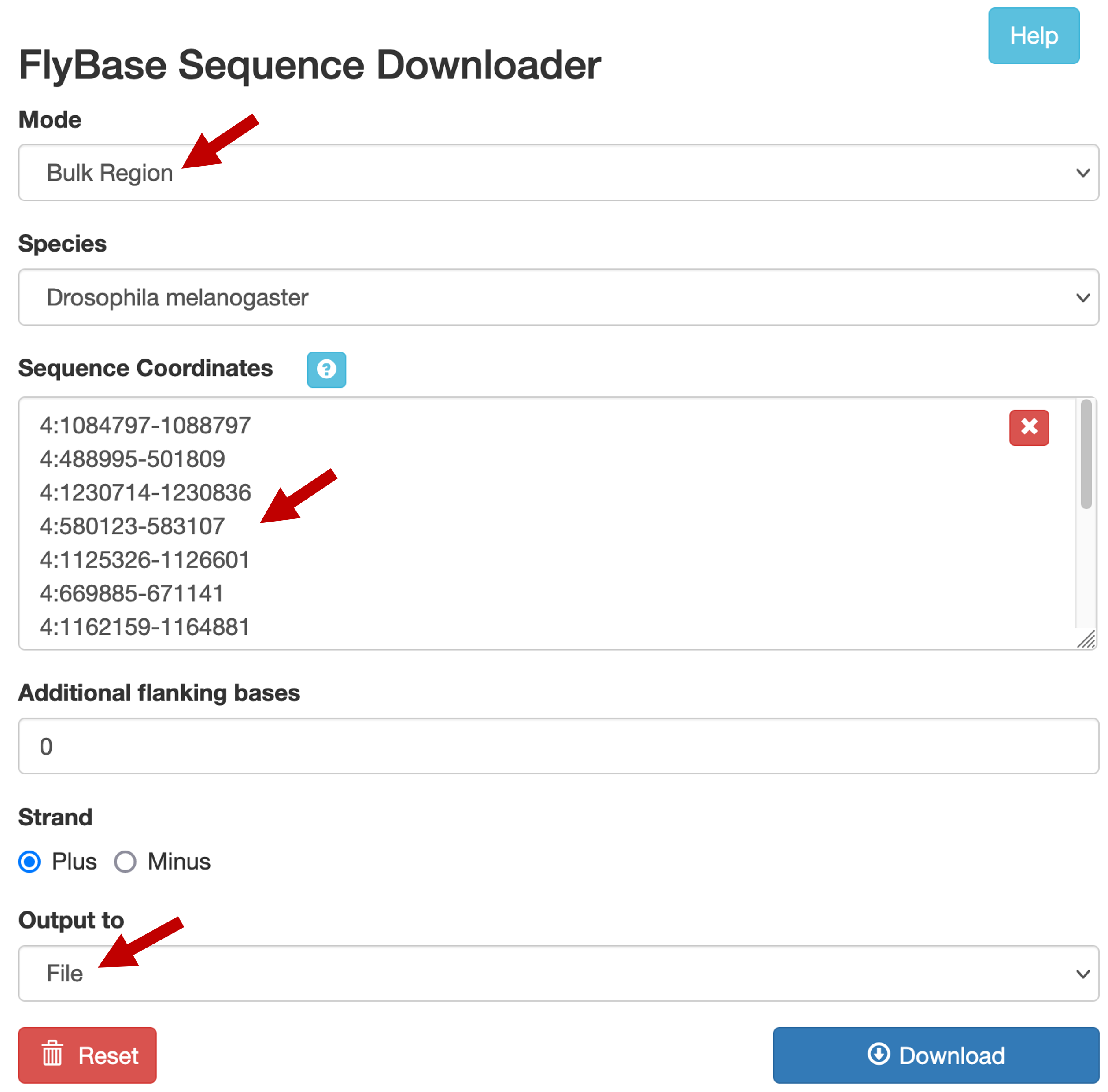
Select the "Bulk Region" option under the "Mode" field. Verify that "Drosophila melanogaster" is selected under the "Species" field. Copy and paste the list of upstream sequence coordinates into the "Sequence Coordinates" text box (Figure 12). Verify that the "Additional flanking bases" field is set to "0", and that the "Strand" field is set to "Plus". Change the "Output to" field to "File". Click on the "Download" button to run the program. Save the output file as dot_genes_head_expressed.fasta.
|
|
If a blank page appears after you click on the "Download" button, you might need to change the configuration of your web browser to allow pop-up windows from FlyBase. Alternatively, you can use the dot_genes_head_expressed.fasta file in the package for this walkthrough. |
To facilitate interpretation of the results during the motif discovery analysis, we will modify the definition lines (i.e., the lines that begin with the > symbol) in the FASTA file. Open the FASTA file in a text editor (e.g., WordPad on MS Windows, TextEdit on macOS). Change each definition line so that it begins with the gene name, followed by a space, followed by "loc=" and the coordinates of the extracted region.
For example, for the upstream region of Actbeta (i.e., the feature at 1084797-1088797), we will change the definition line to the following:
Once you have modified the definition lines, save and close the sequence file.
Identify enriched motifs in the collection of upstream sequences
Now that we have generated the collection of upstream sequences using the FlyBase Sequence Downloader tool, we can search for motifs that are enriched in this collection of sequences. Among the plethora of motif discovery tools that are publicly available, the MEME suite is one of the most popular solutions [Bailey et al., 2009]. In this walkthrough, we will use the web-based MEME interface to analyze our collection of sequences. Navigate to the MEME website, and click on the "MEME" icon (Figure 13).
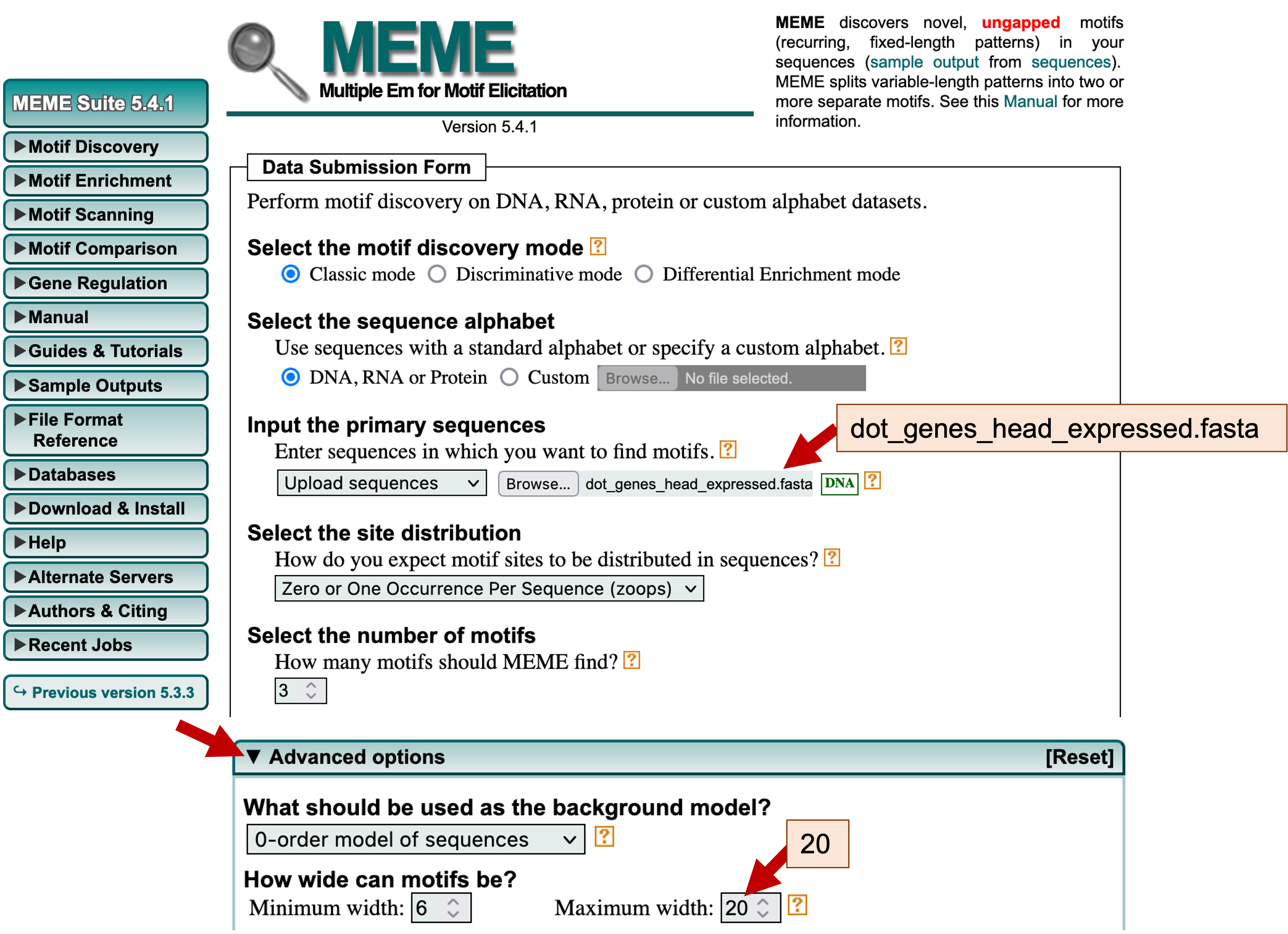
We will perform the initial MEME search using mostly default parameters. Verify that the "Select the motif discovery mode" field is set to "Classic mode." Under the "Input the primary sequences" field, click on the "Browse" or the "Choose File" button and select the file with the upstream sequences (i.e., dot_genes_head_expressed.fasta). Because our collection of upstream sequences may contain different motifs, we should verify that the "Select the site distribution" field is set to "Zero or One Occurrence Per Sequence (zoops)." We should also verify that that the "Select the number of motifs" field is set to 3.
Because most conserved motifs are short (e.g., the average length of transcription factor binding sites is 10 nucleotides [Stewart et al., 2012]), we should also change the maximum width of the sequence motifs that are identified by MEME. Click on the "Advanced options" header to expand this section. Under the "How wide can motifs be?" section, change the "Maximum width" to 20 (Figure 14).
|
|
If a "MEME SUITE NEWS" message box appears at the bottom of the page, close the message box to access the "Advanced options" header. |
Click on the "Start Search" button at the bottom of the page to run the MEME program on our collection of upstream sequences. Depending on how busy the server is, this search could take a while to complete (Figure 15).

This page will refresh automatically when the MEME search is complete. For teaching purposes, the MEME search results (dot_head_expressed_MEME.html) are available inside the MEME_results folder in the package for this walkthrough.
Interpreting the MEME and MAST results
Once the MEME search is complete, the search results page will show links to the MEME and MAST output in three different formats (HTML, XML, and text, Figure 16). The MEME program attempts to find over-represented motifs in our collection of sequences. Once these motifs have been identified, the distributions of these motifs in each upstream sequence are determined by MAST.
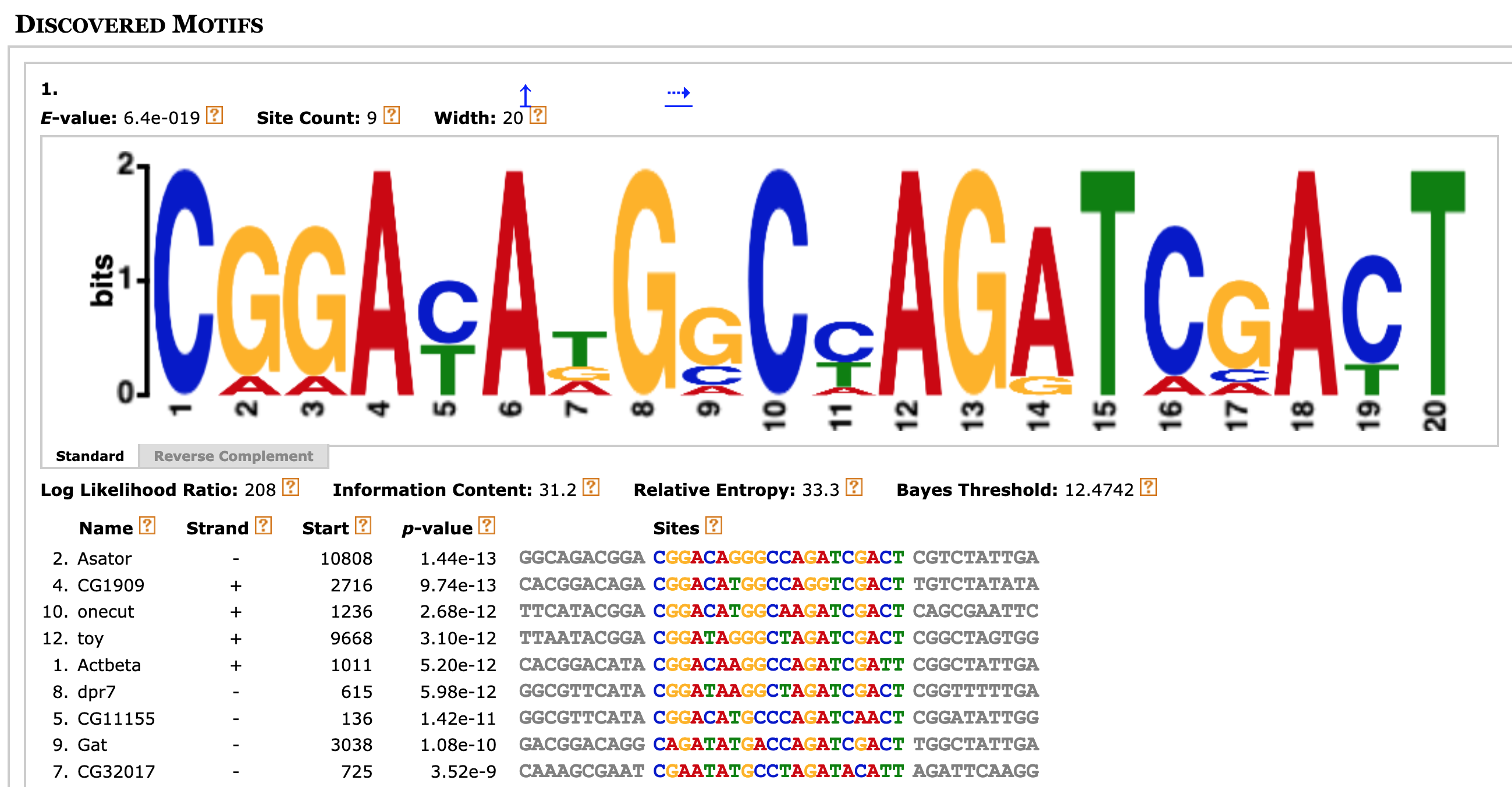
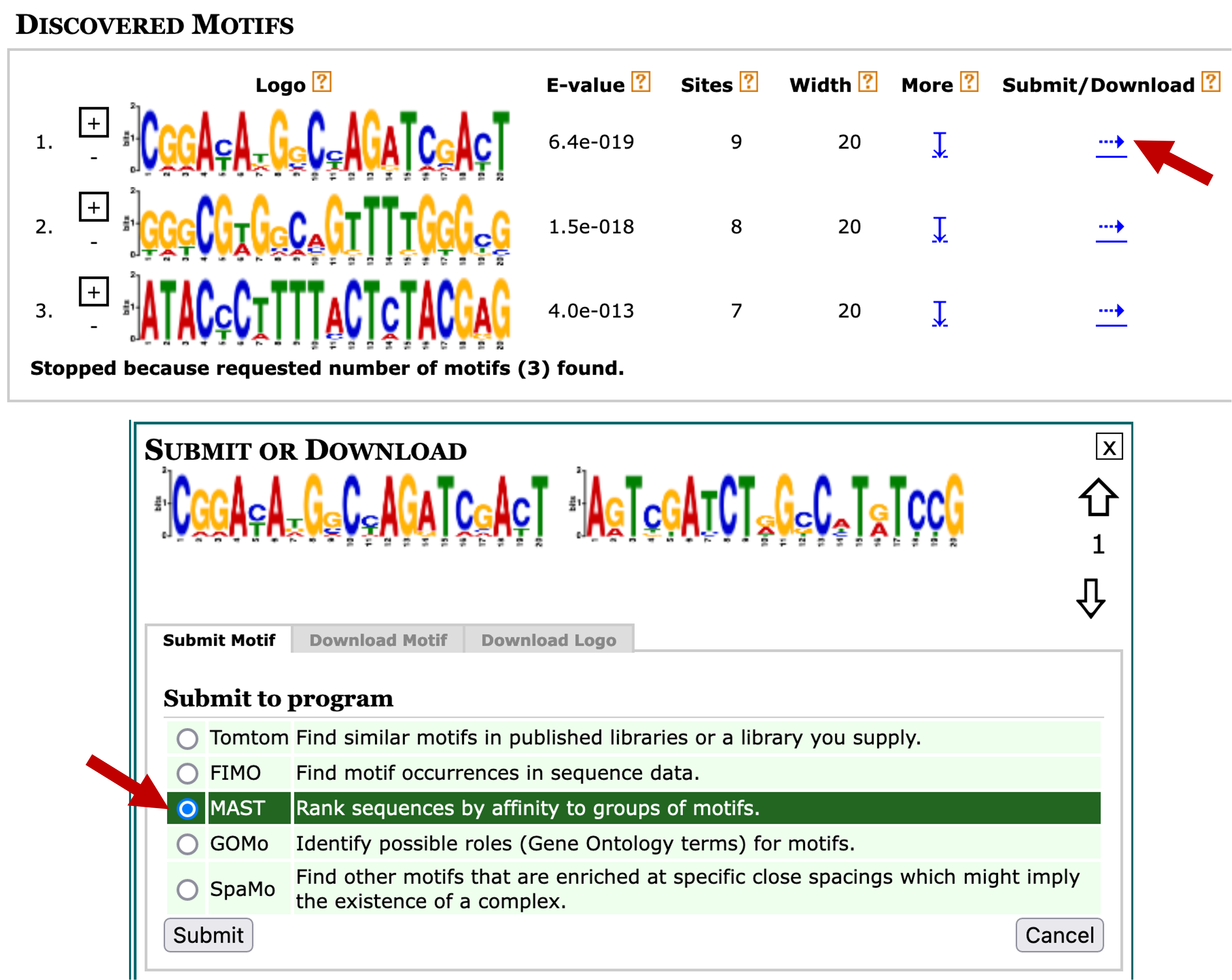
Right click (control-click on macOS) on the "MEME HTML output" link and open the MEME search results page in a new tab. The "Discovered Motifs" section shows the three most significant motifs that were found by MEME (Figure 17). The "Logo" column contains the sequence logo for each motif, where the height of each base corresponds to the level of sequence conservation. The "+" and "-" buttons next to each sequence logo allow us to view the motif in the forward ("+") and the reverse complement ("-") orientation, respectively. The value in the "E-value" column corresponds to the statistical significance of the motif and the value in the "Sites" column corresponds to the number of sites within the primary sequences that were used to construct each motif.

To learn more about the first motif, click on the blue down arrow link under the "More" column (Figure 17). The multiple sequence alignment beneath the sequence logo corresponds to the nine sites that were used to construct the first motif (Figure 18).
Because transcription factors often work in concert with each other, we often would like to know the distribution of all the motifs identified by MEME. A diagram of the collection of non-overlapping motif instances are shown in the "Motif Locations" section along with their statistical significance (height of the block) (Figure 19). A tooltip will appear with additional details on each motif site when you hover the mouse on top of each block.
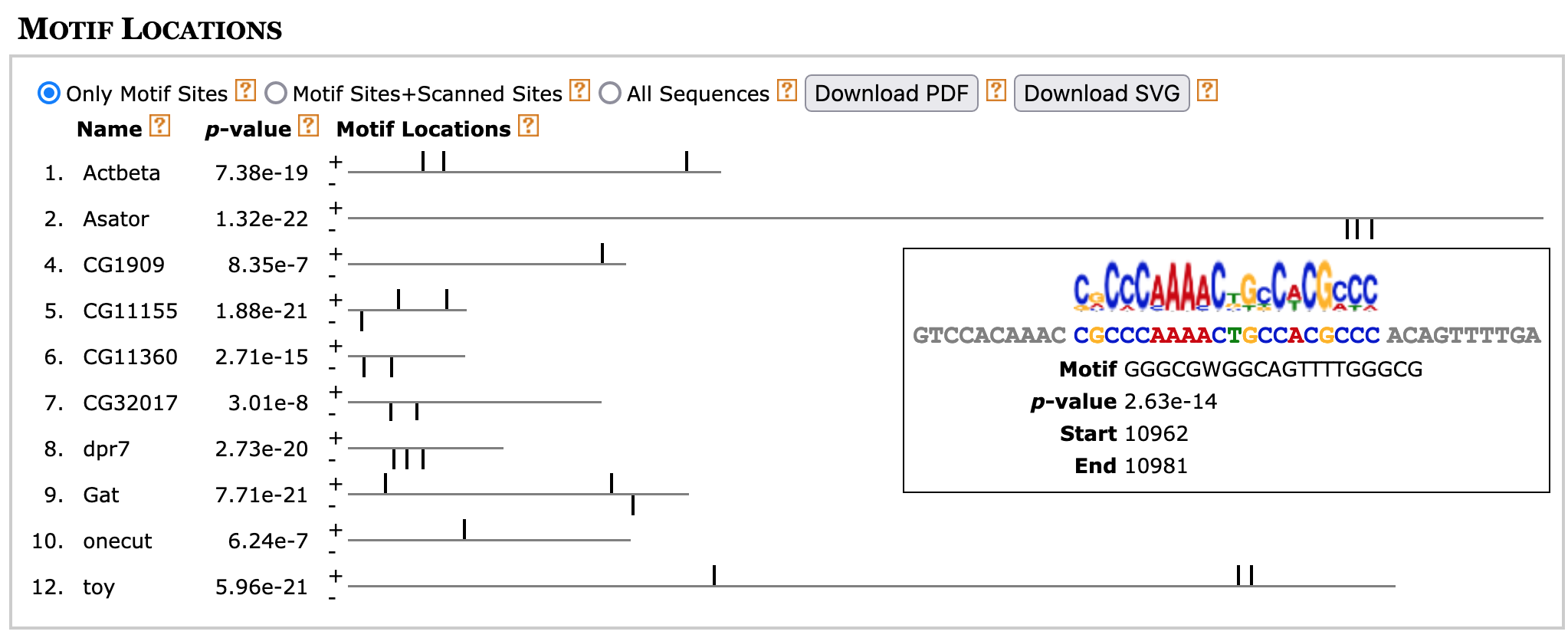
By default, the "Only Motif Sites" option (located underneath the "Motif Locations" header) is selected and the diagram will only show the motif instances that were used to construct each motif. Because we have previously specified in our search parameters that we expect to find zero or one instance of each motif within each sequence, each sequence in the block diagram will contain at most one motif instance.
If you select the "Motif Sites+Scanned Sites" option, MEME will search each motif against the entire collection of upstream sequences and there could be multiple significant matches to each motif. Selecting the "All Sequences" option will show all 12 sequences that are in our input sequence file irrespective of whether they contain any of the three motifs.
We can examine the set of significant non-overlapping motif instances in more detail using the "Top Scoring Sequences" section of the MAST HTML output. To view the MAST results, go back to the tab with the links to the MEME and MAST output (Figure 16). Right click (or control-click on macOS) on the "MAST HTML output" link and open the MAST search results page in a new tab.
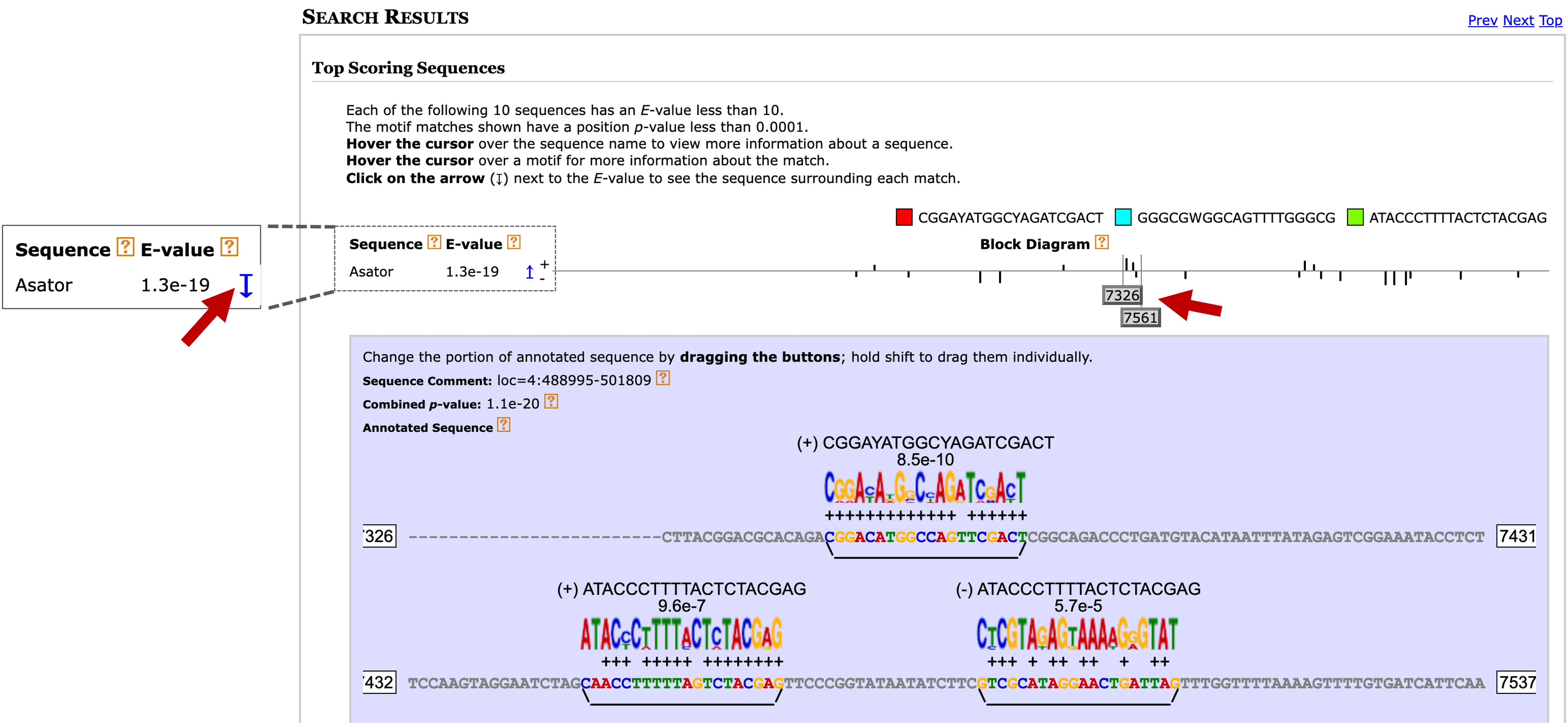
Scroll down to the "Search Results" section. Click on the blue arrow next to the E-value of the sequence you want to investigate (e.g., Asator) to expand the section. Then drag the gray buttons under the Block Diagram section to navigate to the specific motif matches you want to examine (Figure 20). Hold the shift key before dragging the gray buttons in the block diagram to adjust the range of the expanded section.
In addition to searching for motif instances against our collection of upstream sequences, we can also use the MAST tool to search the motifs against other collection of sequences. Go back to the web browser tab with the MEME search results. Click on the blue right arrow under the "Submit/Download" column for motif 1. Select "MAST" under the "Submit to program" section and then click on the "Submit" button (Figure 21).
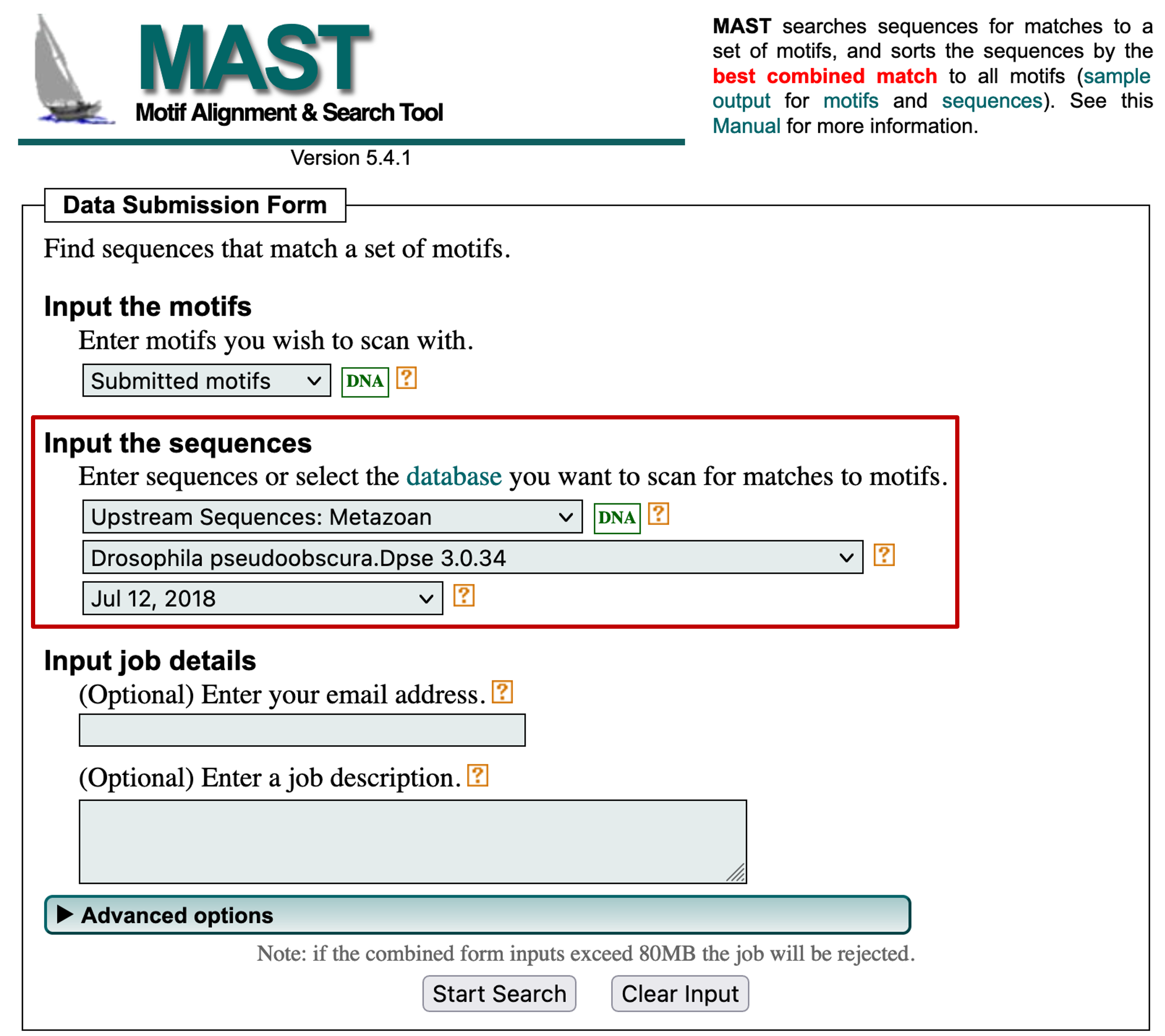
For example, we can use MAST to search the discovered motifs against the collection of sequences upstream of the TSS of D. pseudoobscura genes by changing the "Input the sequences" field to "Upstream Sequences: Metazoan" and then selecting the "Drosophila pseudoobscura.Dpse 3.0.34" and "Jul 12, 2018" options (Figure 22). This search allows us to determine if a motif on the D. melanogaster dot chromosome is also found in D. pseudoobscura. Click on the "Start Search" button to launch the MAST analysis.
Once the MAST search is complete, right click (or control-click on macOS) on the "MAST HTML output" link and open the MAST search results page in a new tab. The "Search Results" section shows MAST has detected 38 D. pseudoobscura upstream sequences with significant (E-value < 10) matches to the first motif discovered by MEME (Figure 23).

The "Sequence" column shows the accession number and the name of the gene that contains the motif. The first part of the name (e.g., FBpp0278184) corresponds to the FlyBase ID and the second part of the name (e.g., GA24903) corresponds to the FlyBase annotation symbol.
In addition to determining the frequency and distributions of the motifs, we can also determine if any of the motifs identified by MEME are similar to known motifs in Drosophila using the Tomtom program [Gupta et al., 2007]. Go back to the MEME results page and then click on the blue right arrow under the "Submit/Download" column for motif 1. Select "Tomtom" under the "Submit to program" section and then click on the "Submit" button (Figure 24).
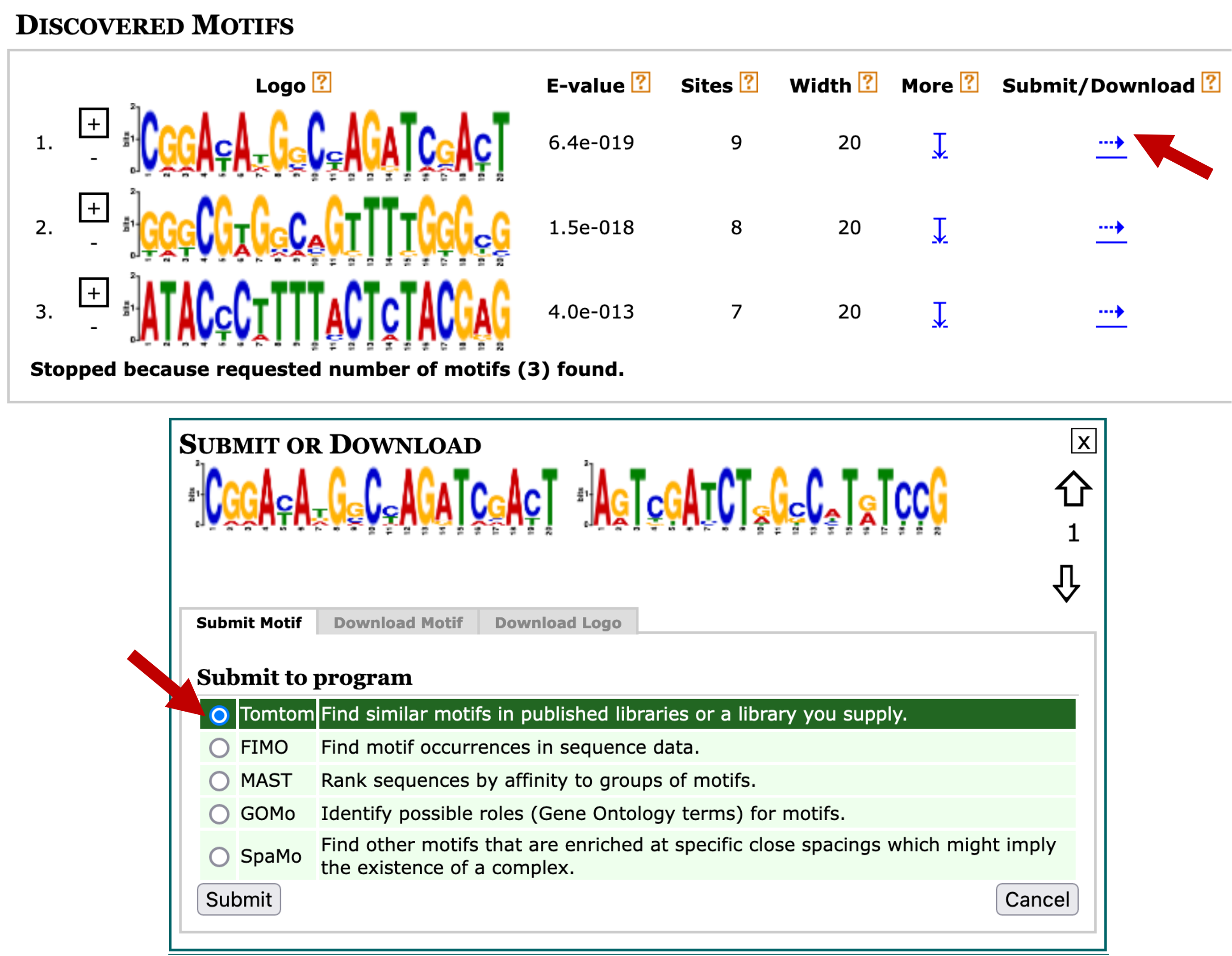
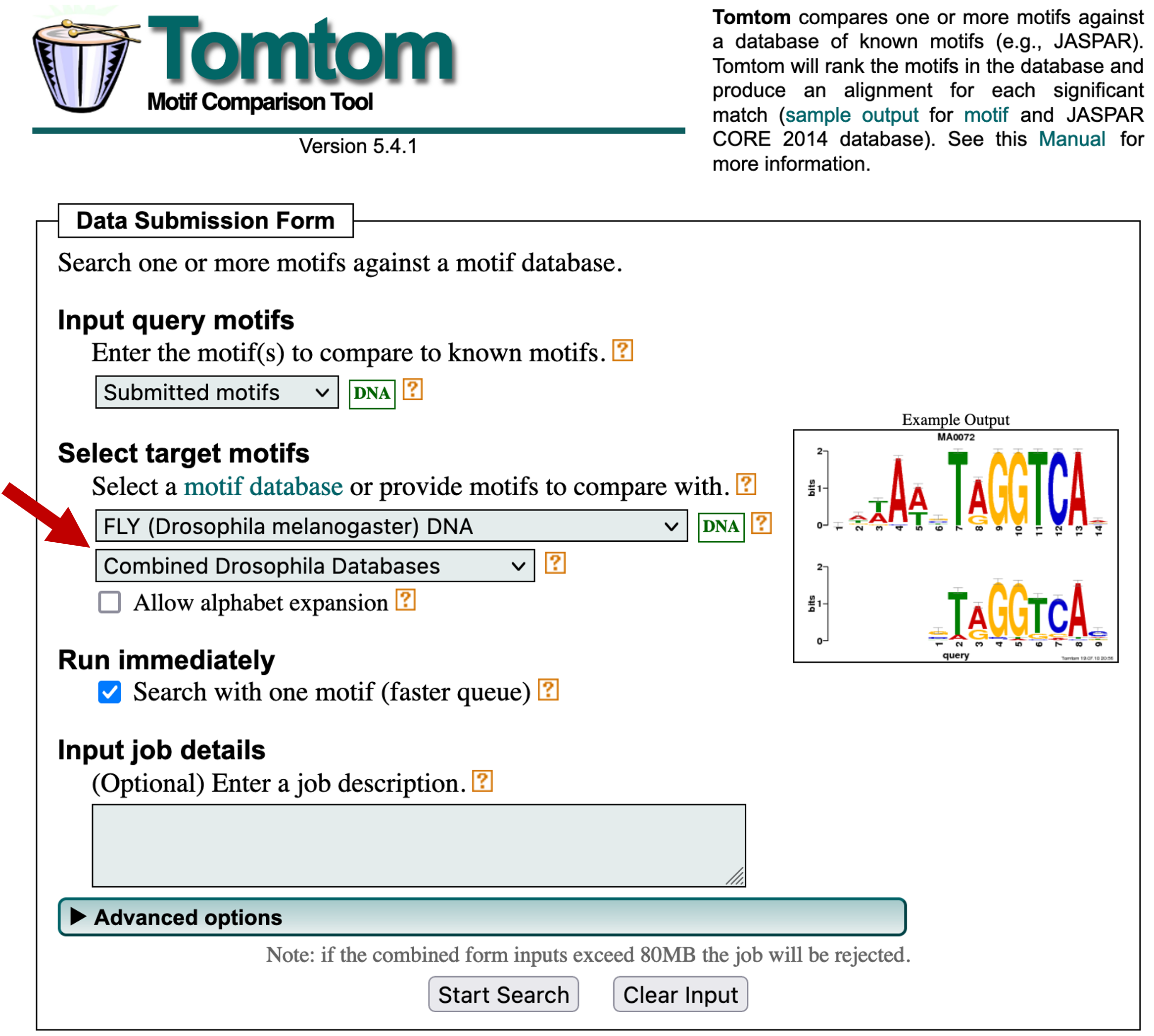
In this example, we will search the first motif identified by MEME against multiple databases of known motifs in D. melanogaster. Select "FLY (Drosophila melanogaster) DNA" and "Combined Drosophila Databases" under the "Select target motifs" field (Figure 25). Click on the "Start Search" button.
Using this configuration, Tomtom will search our motif against five motif databases (OnTheFly_2014, Fly Factor Survey, FLYREG, iDMMPMM, and DMMPMM). These databases contain motifs that are supported by different types of experimental evidence. For example, the transcription factor binding sites in the Fly Factor Survey database were identified primarily using the bacterial one-hybrid (B1H) system [Zhu et al., 2011].
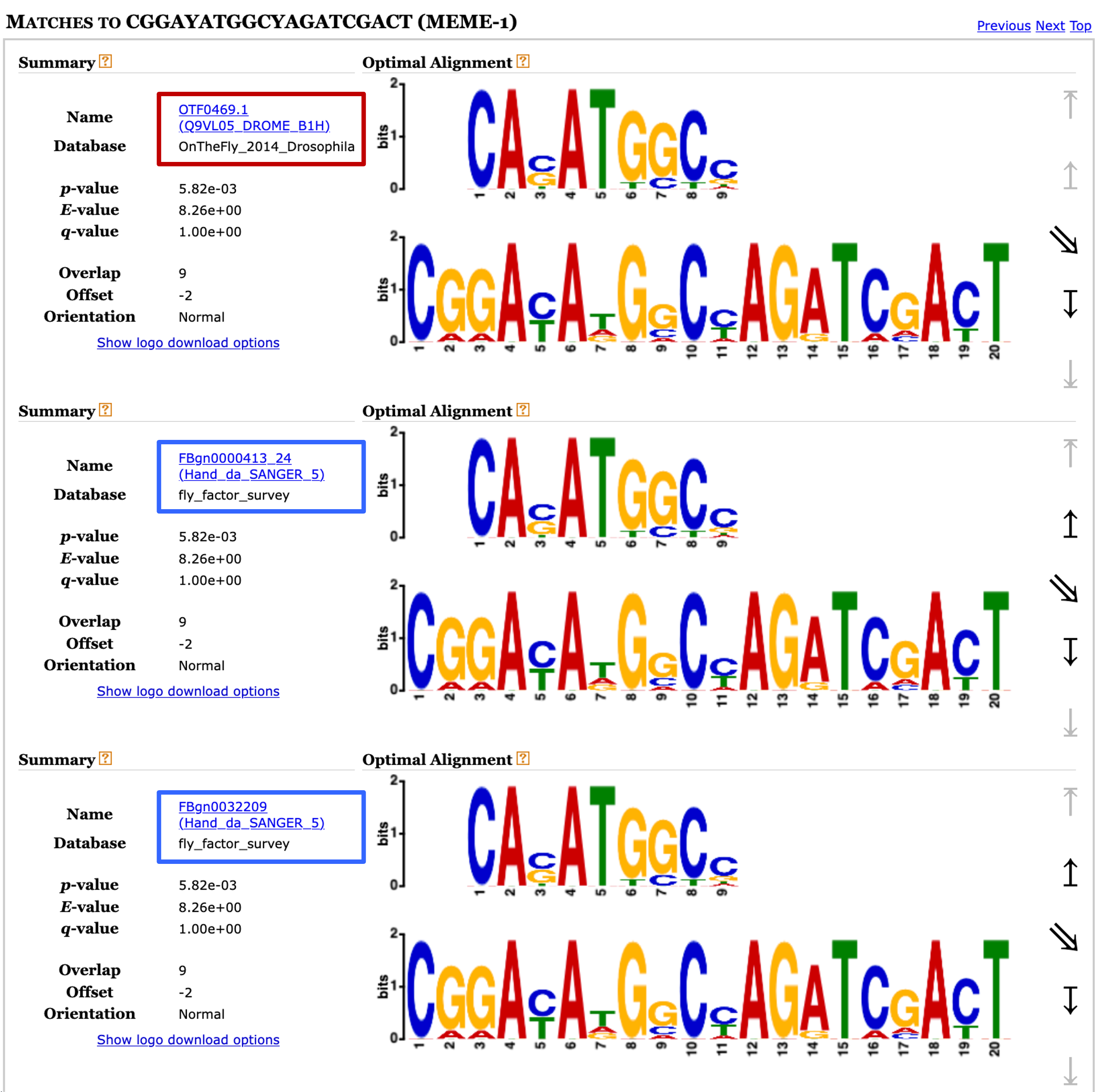
This Tomtom search might take a few minutes to complete. Once the results are available, click on the "Tomtom HTML output" link to view the search result. We can see under the Query Motifs section that Tomtom has identified five matches to motif 1 (Figure 26). A closer examination of the list of matches shows that two of the matches (FBgn0000413_24 and FBgn0032209) have the same alternate name (Hand_da_SANGER_5), indicating that these two matches refer to the same motif record.

Examination of the "Target Databases" section shows that three of the matches are to motifs in the Fly Factor Survey database and two of the matches are to motifs in the "OnTheFly_2014_Drosophila" database. The sequence logos under the "Matches to 1" section shows that two of the motif matches from the Fly Factor Survey database (FBgn0000413_24 and FBgn0032209) are identical.
In addition, the two motif matches to the "OnTheFly_2014_Drosophila" database are identical to the motif matches to the Fly Factor Survey database. For example, the sequence logo for the motif OTF0469.1 in the "OnTheFly_2014_Drosophila" database is the same as the FBgn0000413_24 and FBgn0032209 motifs in the Fly Factor Survey database (Figure 27). Similarly, the sequence logo for the "OnTheFly_2014_Drosophila" motif OTF0489.1 is identical to the Fly Factor Survey motif FBgn0035454. Hence there are only two distinct motifs in the Tomtom search results.
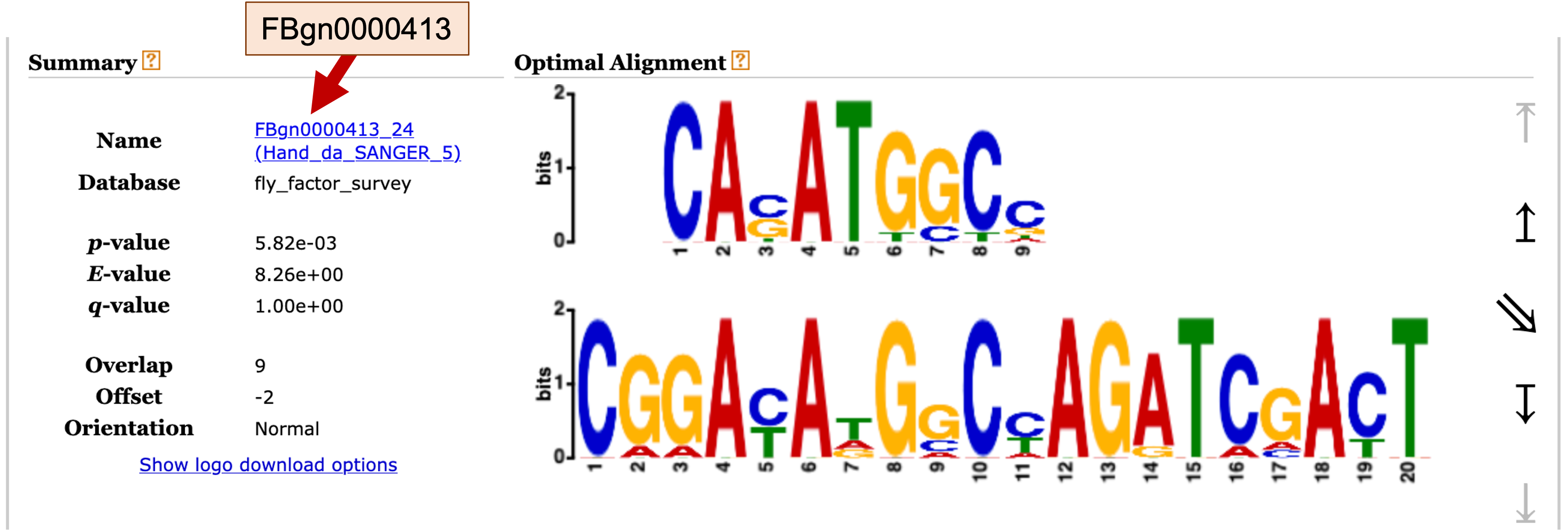
For motif records in the Fly Factor Survey database, the name of the motif includes the FlyBase ID for the gene associated with the motif. For example, for the Fly Factor Survey motif match FBgn0000413_24, the part of the name before the underscore (FBgn0000413) corresponds to the FlyBase gene ID (Figure 28).
To learn more about the gene associated with this motif, open a new web browser tab and navigate to FlyBase. Enter "FBgn0000413" into the "Jump to Gene" (J2G) search box on the top right corner of the navigation bar, and then click "Go". On the HitList page, select "D. melanogaster" under the "Filter by species" panel, and select "Gene" under the "Filter by data class" panel. Click on the "da" button to navigate to the gene record (Figure 29).
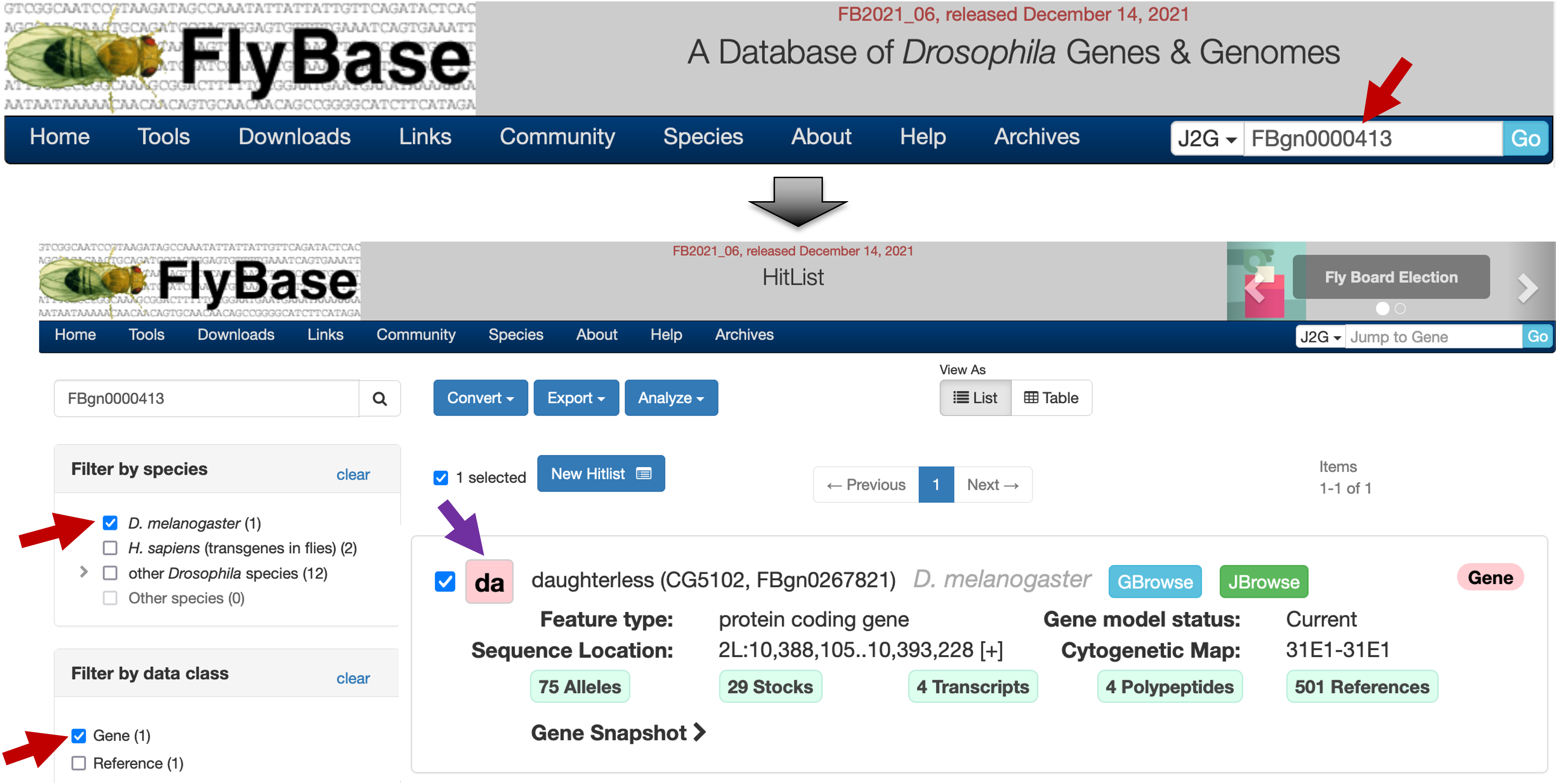
The Gene Summary section of the FlyBase Gene Report for da indicates that the gene encodes a protein with a basic helix-loop-helix (bHLH) domain. This gene is involved in sex determination, dosage compensation, and in the transcriptional regulation of oogenesis, neurogenesis, myogenesis and cell proliferation (Figure 30).
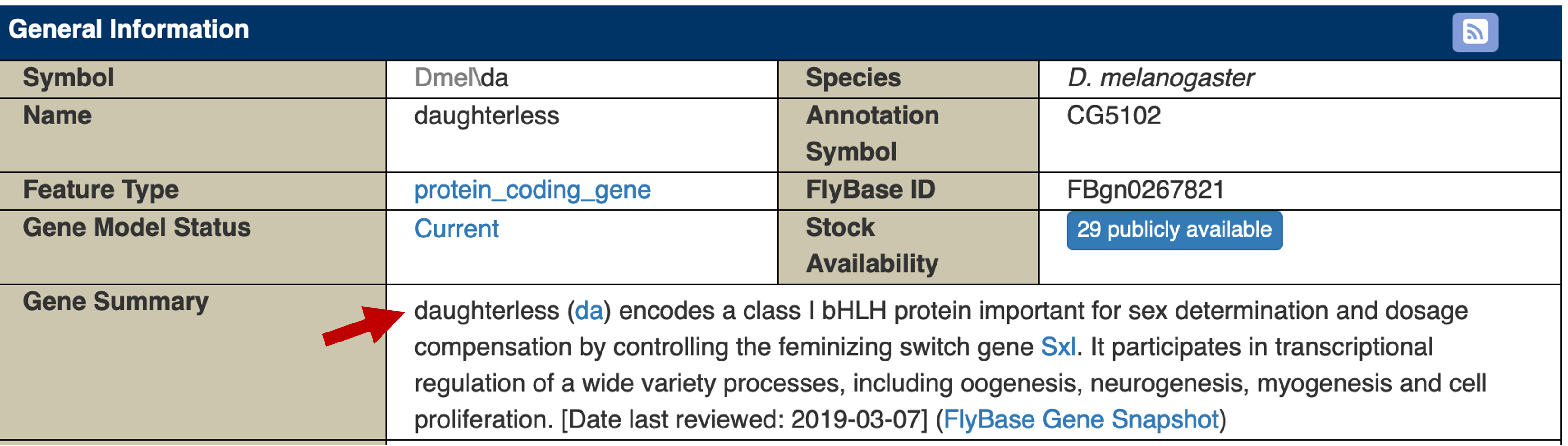
To determine the list of proteins that are known to interact with Daughterless, click on the "Interactions" link on the "Report Sections" sidebar. The esyN Network Diagram under the "Summary of Physical interactions" section shows that Daughterless has known physical interactions with multiple proteins. One of the proteins which interact with the Daughterless protein is Hand (Figure 31).
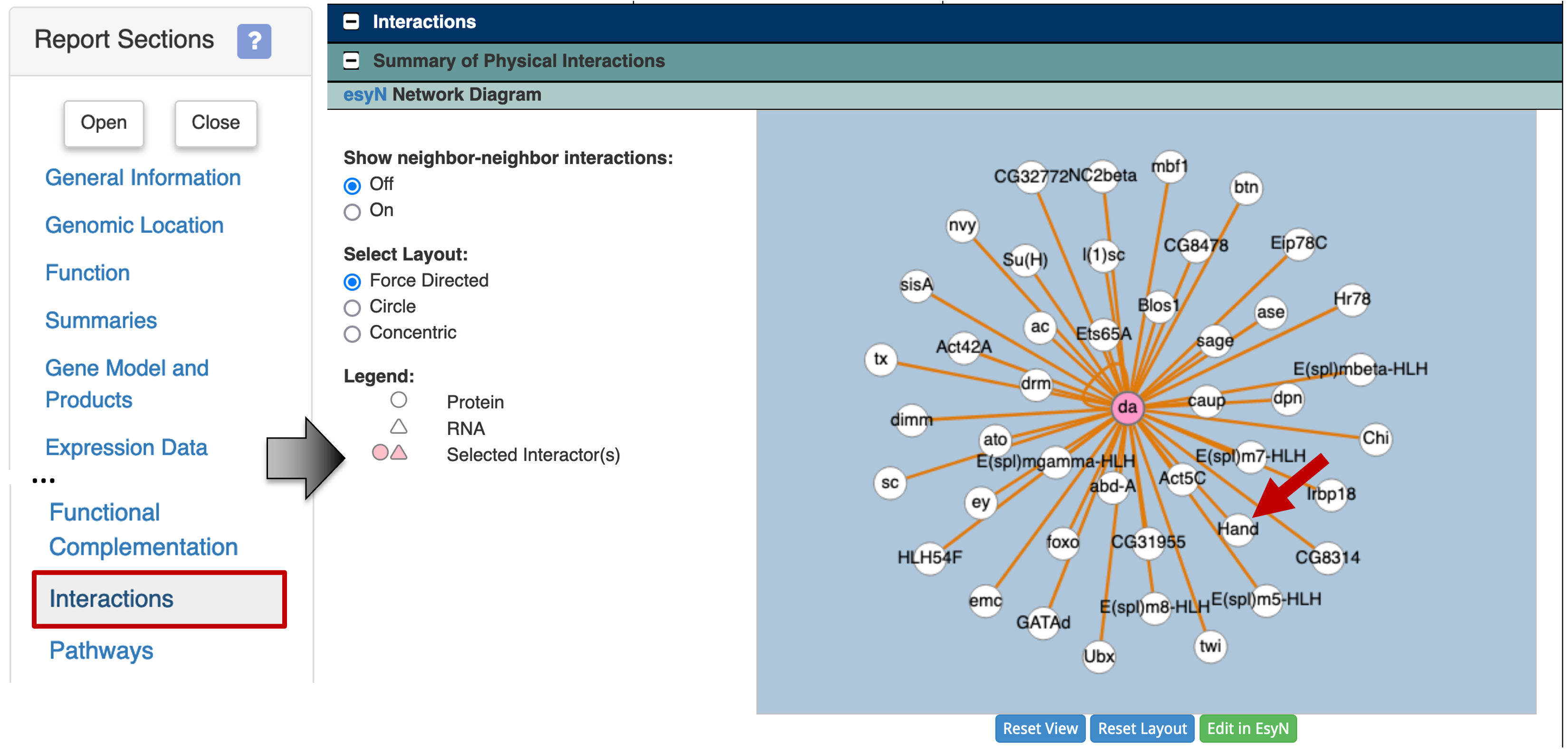
To learn more about the interaction between Daughterless and Hand, scroll down to the table under the "protein-protein" section (Figure 32). The table shows that the physical interaction between Daughterless and Hand (da - Hand) was identified using the two hybrid assay by Tögel and colleagues ([Tögel et al., 2013]; FBrf0222537). This study suggests that Daughterless and Hand are dimerization partners, and they are both required for the development of Drosophila wing hearts.
This explains why the motif identified by MEME has the label Hand_da_Sanger_5, and matches to both FBgn0000413_24 and FBgn0032209 in the Tomtom search result. The FlyBase ID for the Hand gene is FBgn0032209.
We can apply the same approach to investigate the other motif match [FBgn0035454 (CG12029_SANGER_10)] identified by the Tomtom search. This investigation is left as an exercise for the reader.
Conclusions
This walkthrough illustrates the use of multiple web databases and tools to identify conserved motifs in a collection of sequences upstream of the transcription start site. Using the FlyBase RNA-Seq Expression Profile Search tool, we identified a set of genes with similar expression patterns. Using the FlyBase search results table, we can apply additional filters to this list of genes (e.g., by focusing on only dot chromosome genes or genes with a particular gene ontology term). In addition, we can export the gene list in many different formats for downstream analyses.
We then used the FlyBase Gene Report and JBrowse to determine the coordinates of the regions upstream of the TSS. We can retrieve the genomic sequences for these upstream regions using the FlyBase Sequence Downloader tool.
Running MEME on this collection of upstream sequences allow us to identify over-represented motifs. We can examine the distribution of the motif instances using the MAST tool. To characterize this set of motifs, we used Tomtom to compare the motifs discovered by MEME against multiple databases of known motifs in Drosophila. For MEME motifs that match known motifs in the motif databases (e.g., Fly Factor Survey), we can further investigate the set of proteins (e.g., transcription factors) that are associated with these specific binding sites.
Bibliography
-
[Bailey et al., 2009] Bailey, T. L., M. Boden, F. A. Buske, M. Frith, C. E. Grant et al., 2009 MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37: W202–208.
-
[Graveley et al., 2011] Graveley, B. R., A. N. Brooks, J. W. Carlson, M. O. Duff, J. M. Landolin et al., 2011 The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479.
-
[Gupta et al., 2007] Gupta, S., J. A. Stamatoyannopoulos, T. L. Bailey, and W. S. Noble, 2007 Quantifying similarity between motifs. Genome Biol. 8: R24.
-
[Stewart et al., 2012] Stewart, A. J., S. Hannenhalli, and J. B. Plotkin, 2012 Why transcription factor binding sites are ten nucleotides long. Genetics 192: 973–985.
-
[Tögel et al., 2013] Tögel, M., H. Meyer, C. Lehmacher, J. J. Heinisch, G. Pass et al., 2013 The bHLH transcription factor hand is required for proper wing heart formation in Drosophila. Dev. Biol. 381: 446–459.
-
[Zhu et al., 2011] Zhu, L. J., R. G. Christensen, M. Kazemian, C. J. Hull, M. S. Enuameh et al., 2011 FlyFactorSurvey: a database of Drosophila transcription factor binding specificities determined using the bacterial one-hybrid system. Nucleic Acids Res. 39: D111–117.